
英语原文共 11 页,剩余内容已隐藏,支付完成后下载完整资料
热固性树脂固化收缩率测量技术评估
摘要:树脂化学收缩决定了复合结构的表面完整性和粗糙度。因此,为了减少表面失效并产生良好的表面质量,在固化过程中能够测量和跟踪树脂收缩率是必不可少的。该手稿使用流变仪,氦基比重瓶和热机械分析仪(TMA)对环境固化UP和环氧树脂进行实时树脂收缩的测量和监测。采用流变仪最新开发的鲁棒技术获得的收缩读数与传统比重法的读数一致。它们在UP和环氧树脂体系的公认文献值中也分别为7-10%和3.5-4.5%。后固化期间的收缩测量在高温下有效进行,表明所提供的方法可以应用于非环境固化体系。发现TMA不能可靠地测量收缩率。
关键词:化学收缩 固化行为 比重瓶 流变仪 热分析 热固性树脂
1.前言
1.1固化过程中的化学收缩
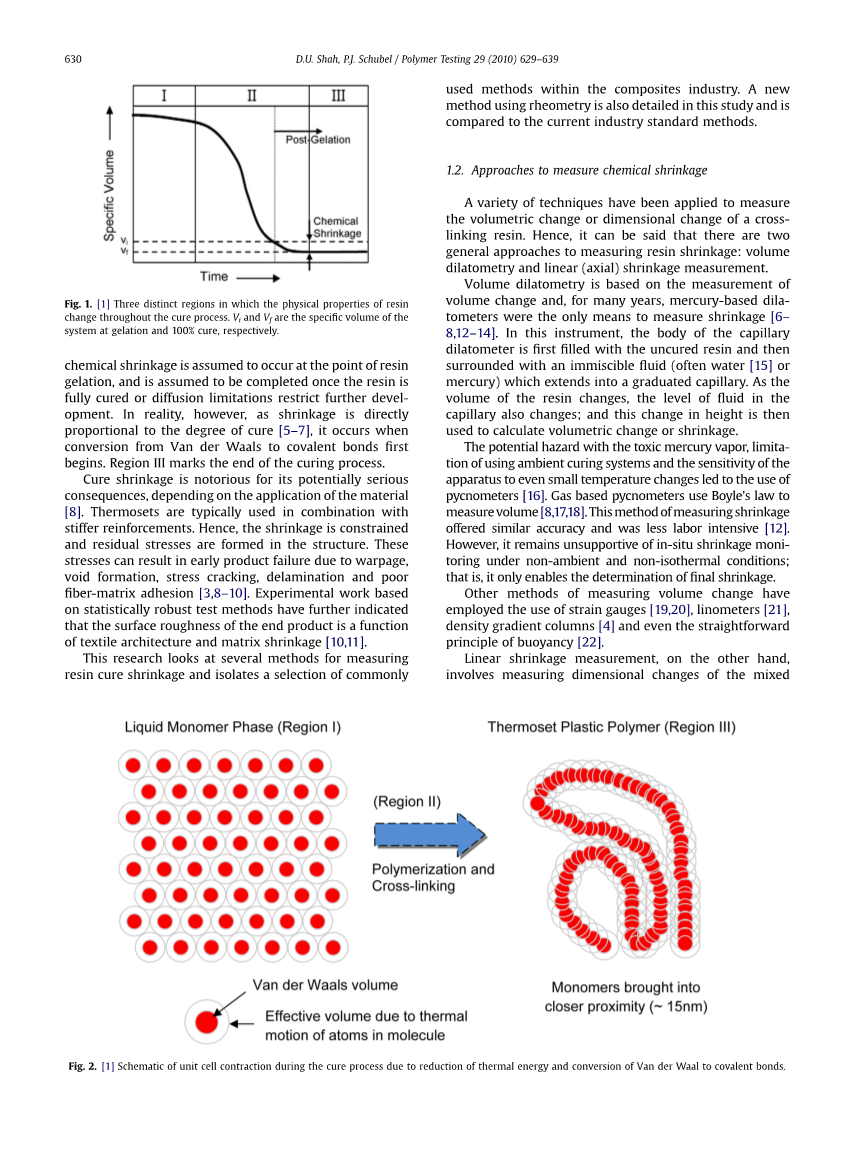
通过能量和/或催化剂活化的固化过程将热固性树脂转化为坚硬的难处理固体(塑料或橡胶)。 固化过程通常通过三个不同的区域来描述(图1)[1]。 在I区,树脂未固化,表现为粘性流体。 液体移动单体分子可以被认为是相互分离的化学实体。 每个单位体积都是由范德瓦尔斯体积和热能决定的(图2)[2]。
区域II表示树脂的固化阶段。在此放热过程中,单体分子首先通过支化(聚合)发生链形成和线状生长[3]。然后由此产生的分子链形成交联,从而形成大的刚性三维分子网络。
聚合和通过支化的附加键合导致储存热能的自由度降低。 这意味着之前的流动和离散单体变得受限且密集的聚合物单元(图2)。 而且,分子之间的范德华键可转化为更强更短的共价键。 然后观察系统的硬度(化学硬化)和特定体积(化学收缩)的显着增加。
如果该系统在高温下固化或经历重要的放热反应并因此需要额外的冷却,则可观察到额外的收缩(热收缩)。 然而,这取决于i)树脂在高温固化过程中的热膨胀,ii)更充分固化的材料的收缩和iii)冷却过程中树脂的收缩[4]。
对于本研究,为了在不压缩初始液态树脂的情况下进行线性收缩测量,假定树脂化学收缩发生在树脂凝胶化点,并且假定一旦树脂完全固化或扩散限制限制了进一步发展,就完成树脂收缩。 然而,实际上,由于收缩率与固化程度成正比[5-7],因此当从范德华转变为共价键首先开始时就会发生收缩。 区域III标志着固化过程的结束。
根据材料的应用[8],固化收缩对其潜在的严重后果而言是非常重大的。 热固材料通常与更坚硬的增强材料结合使用。 因此,收缩受到限制,在结构中形成残余应力。 这些应力可能导致产品早期由于翘曲,空洞形成,应力开裂,分层和纤维基质粘附性差而失效[3,8-10]。 基于统计学上稳健的测试方法的实验工作进一步表明,最终产品的表面粗糙度是纺织结构和基体收缩的函数[10,11]。
本研究着眼于测量树脂固化收缩率的几种方法,并分离出复合材料行业中常用的一些方法。 本研究还详细介绍了一种使用流变测量的新方法,并将其与当前工业标准方法进行了比较。
1.2测定化学收缩的方法
已经应用了各种技术来测量交联树脂的体积变化或尺寸变化。 因此,可以说有两种测量树脂收缩的一般方法:体积膨胀测量和线性(轴向)收缩测量。
体积膨胀法是基于体积变化的测量方法,多年来,基于汞的膨胀计是测量收缩的唯一手段[6-8,12-14]。 在该仪器中,毛细管膨胀计的主体首先填充未固化的树脂,然后用不可混溶的液体(通常是水[15]或汞)包围,并延伸到分级毛细管中。 随着树脂体积的变化,毛细管中的液面也发生变化; 然后使用这种高度变化来计算体积变化或收缩。
由于有毒汞蒸气的潜在危害,使用环境固化系统的限制以及仪器对温度变化的敏感性,研究开始改用比重瓶[16]。 气体比重瓶使用波义耳定律测量体积[8,17,18]。 这种测量收缩率的方法具有相似的精度,并且劳动强度较低[12]。 然而,在非环境和非等温条件下,它仍然不支持原位收缩监测; 也就是说,它只能确定最终的收缩。
其他测量体积变化的方法采用了应变仪[19,20],测角仪[21],密度梯度柱[4],甚至是直接的浮力原理[22]。
另一方面,线性收缩测量涉及测量混合树脂样品的尺寸变化并进行假设,如平面应变条件[10]和各向同性收缩[23],以得出体积收缩的最终值。
现代流变仪可以配置成测量线性收缩率并研究树脂的流变行为[10,24]。 扭矩或应变控制测试不仅可以确定胶凝点,而且可以监测树脂在固化时的尺寸变化。
热机械分析仪(TMA#39;s)曾被用于测量线性尺寸变化,通过测量样品在施加载荷和等温条件或特定温度斜坡下固化时树脂厚度的变化[12]
激光干涉[25],光纤和动态机械分析仪(DMA)[8,26]也被用于测量线性收缩到合理的精度。
本文研究了使用流变仪,氦基比重瓶和TMA测量树脂收缩率。 采用差示扫描量热仪(DSC)测量树脂的固化度(DoC),从而能够比较三种方法对于DoC的实时树脂收缩率。 对三种方法的结果进行全面的比较以评估一般方法的准确性和精确度以及其他优点或缺点。 然后确定测量收缩最有效的方法。
2.方法
2.1材料:树脂选择
表1列出了本研究考虑的树脂体系。环境固化体系是可以期望的,因此可以在比重瓶的整个固化过程中监测收缩率。 不饱和聚酯树脂(UP)系统得自Reichhold Norpol和得自Gurit UK Ltd.的环氧树脂系统
本研究的一个合适的树脂体系是在环境温度(25°C)下固化,具有相当短的固化时间,低到中等的峰值放热温度(lt;70°C)并具有缓慢的初始固化速率。 为了选择最适合的系统,使用DSC在整个三区固化过程中跟踪每个树脂系统的温度曲线(图3)
据观察,环氧体系具有较低的初始固化速率,但花费较长时间才能达到峰值外热量并完全固化。 UP系统迅速达到峰值放热,但具有较高的初始固化速率。 此外,UP系统在固化过程中的环境温度远高于环氧树脂。
图3使我们能够将树脂体系分为两个不同的类别:第一类适用于TMA和流变测定分析,第二类用于比重分析。
I类树脂在少量时间内显示出大的温度变化; 这些不适用于pycno-metric(吡咯烷酮)分析,因为使用比热计的体积测量应在等温条件下进行。
II类由适用于比重计分析的树脂组成,因为它们的初始固化速度较慢,因此可以跟踪体积变化以获得更好的分辨率。 此外,由于这些树脂体系显示较低的峰值温度,因此体积读数不太可能受热收缩影响。
对于该研究,选择UP型420-100(具有0.25wt%的NL 49P促进剂和1wt%的Butanox M50引发剂)和Ampreg 21环氧树脂(具有33wt%的Ampreg 21缓慢固化剂)。 将混合物在25°C下充分混合,注意树脂流变性(包括收缩)对树脂与催化剂的比例以及固化过程中的温度的依赖性。
选择这两个系统是因为它们最符合标准。 另外,来自两个不同类别的树脂允许对各种收缩测量技术的准确性进行真正的交叉比较研究。
2.2实验
2.2.1流变仪
使用Bohlin Instruments C-VOR 200流变仪进行树脂的线性收缩测量和流变测定分析。 所采用的实验装置如图4所示。该试验采用振荡模式下的R20mm平滑铝平行板。 基板刚性固定,而顶板可以垂直移动,因此能够施加压缩法向力Fz,并且传递转矩或旋转频率f,基本上允许剪切应变传递至树脂样品。
为了确定树脂的线性粘弹性响应(LVR)区域,首先进行振荡模式下的幅度扫描。 由于LVR是与应变相关的功能,在应变控制下,扫描在等温条件下为25plusmn;0.2°C进行。 因此,在0.2Hz的恒定频率下,在一定范围的特定角度应变幅度(0.003%-30%)下测量样品的VR。 间隙设定为0.5毫米。
在确定的线性区域(材料性质恒定)下,然后进行振动模式下的应变控制频率扫描以研究作为频率函数的树脂的粘弹性性质。 这是通过在0.003%的恒定角度应变下在特定频率范围(0.2 Hz-30 Hz)测量树脂的VR来完成的。 间隙设定为0.5毫米。
振幅和频率扫描表明树脂保持在线性区域的应变和频率范围。 这使参数可以为实验的其余部分设置。 主要测试程序包括三部分运行流变仪:
●细分I-预胶凝
●分段II-后胶凝
●第三部分 - 后固化
在I阶段中,振荡模式以0.2Hz进行单频率应力控制测试,板之间的间隙保持恒定在0.5mm并且应变设定在15%。 当树脂处于液体状态时,不施加正常的力。
在凝胶化时,测试被移到第二部分。 对于本研究,弹性模量G0和粘性模量G00的交叉点被认为是树脂的液 - 固转变点,因为Grsquo;现在可与Grsquo;rsquo;相近。
在II阶段中,树脂处于液 - 固转变点。 单频应力控制测试在振荡模式下以30 Hz进行。 在等温环境条件下施加500mNm的扭矩和0.1N的恒定压缩法向力。 设备设置为补偿间隙以保持压缩法向力恒定在0.1N。将实际间隙与时间作图以监测固化过程中的线性尺寸变化。 由于凝胶化的树脂和施加的低法向力,没有材料从板之间被挤出,从而给出错误的测量结果。
一旦间隙停止变化,流变仪将在第三部分中运行。 使用扩展温度控制(ETC)烘箱在55°C 0.2°C下对树脂进行后固化分析6小时。 第二部分的所有其他条件仍然有效,而实际的差距则受到监控。 6小时后,再次在环境温度下跟踪间隙以消除热收缩的影响并获得实际的化学收缩。
最后,间隙的变化用于使用等式(1)计算树脂化学收缩εv,其中h0是初始间隙,h是实际间隙。 这个方程源自于M. Haider et al。 假设树脂样品的面内应变为零。 这个假设可以通过对板中样品的视觉检查来证实,因为树脂和板之间的强粘附力防止了平面内的运动。 进一步的假设是树脂是不可压缩的。 εv的负号表示体积收缩。
(1)
2.2.2比重瓶
校准的Micromeritics AccuPyc 1330气体分析仪用于测量和监测树脂样品在环境温度下固化时的体积。 该仪器通过测量排出的气体量来工作(图5)。 填充样品室后观察到的压力,然后将其抽空到第二个空的膨胀室中,可以计算样品的固相体积。 最后的体积读数是五次净化和运行的系统读数的平均值。
对于这个测试,首先将空的一次性铝杯放置在样品室中,并测量其体积Vc。 然后,将大约4cmsup3;的未固化树脂倒入杯中,通过从总体积中减去Vc获得未固化树脂Vi的体积。 随着树脂固化(段I),在环境条件下以相当规则的间隔对样品体积Vf进行测量。
通过将树脂填充的杯放入烘箱中,在55℃下进行后固化(段III)6小时。 由于比重瓶对环境体积测量的限制,只有在第III部分完成后才能测量最终的体积读数。
为了确定在固化过程中在时间t的树脂化学收缩εv,使用等式(2)。 同样,εv的负号表示体积收缩。
(2)
2.2.3热机械分析仪
TA Instruments TMA Q400也用于通过H. Yu等人采用的方法测定UP和环氧树脂体系的线性收缩率。[12]。 使用的测试台如图6所示。一次性显微镜载片(10mmtimes;10mm见方)放置在台架屏蔽上,其厚度T在25°C测量。 然后将0.005cmsup3;的混合树脂置于底部显微镜载玻片上并用顶部载玻片覆盖。 通过将两个载玻片摩擦在一起形成约20mm厚的树脂层。 通过测量夹心样品的总厚度来确定树脂层的初始厚度h0。 因此,尺寸变化(h0-h)或新树脂厚度h的测量在25°C等温固化下进行直至固化完成。 也施加了0.02N的非常低的恒定压缩载荷。 可以使用等式(1)计算在时间t时树脂的化学收缩。
2.2.4差示扫描量热仪(DSC)
使用TA Instruments DSC Q10在固化过程中定期测量树脂体系的固化度(DoC)。 在扫描温度模式下,从20°C到200°C以10°C / min的速率进行动态分析,测试了大约20毫克的混合树脂。
使用通用分析软件计算每个样品在固化过程的时间t时的反应的特定热焓Delta;Ht。 为此,假定线性或曲线近似,每个图上的起始点由软件确定并手动接受为执行数值积分的极限。 然后用等式(3)确定DSC中树脂固化(DoC)的分数。
(3)
其中,Delta;Ht是树脂固化过程中t时刻的比热容,Delta;Ho是t = 0分钟时的参比比热容。
3.结果与讨论
3.1不饱和聚酯树脂
3.1.1流变仪
初始凝胶点测定试验和流变仪上UP分段I测试揭示了一系列Grsquo;和Grsquo;rsquo;特征曲线,其中凝胶化时间由Grsquo;和Grsquo;rsquo;交叉点给出。
表2显示了凝胶化时间和Grsquo;(和Grsquo;rsquo;)在多次运行中的相关值。该UP系统的平均凝胶化时间评估为1733s(28.9min),范围约为150s(2.5分钟)。 小范围突出了使用流变仪的UP系统的凝胶点测量精度,注意到在树脂混合后样品不会同时放在铝板上,所以这些值会存在固有差异。 还观察到G0和G00每次(14.7Pa)以几乎相同的值交叉,因此进一步说明所用方法的可靠性。
第II部分显示随着平行板之间间隙变化测量的DoC增加,树脂收缩的进展。 在第二部分中进行三次重复测试(图7中显示的两个测试)约20小时,之后未检测到间隙变化。 三次测试产生的结果在性质上非常相似,并显示最终间隙尺寸为0.453 mmtimes;0.001 mm。 在第二部分结束时,UP系统显示化学收缩率为9.17%,这在文献值7-10%范围内[9,10]。
试验2初期间隙的大小有明显波动,试验3的大约630
全文共14646字,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[10662],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- BaTiO3和SrTiO3纳米立方单晶体的有 序组装的压阻响应特性外文翻译资料
- 结构对有机硅改性酚醛树脂热稳定性及抗氧化机理的影响外文翻译资料
- 磷酸三(2-巯基乙基)固化环氧热固树脂的高折射率和阻燃性外文翻译资料
- 燃烧合成TiB2-Cu金属陶瓷的抗烧蚀性外文翻译资料
- 氢键在光诱导水离解中的作用:一把双刃剑外文翻译资料
- 碳酸氢钠/偶氮二异丁腈协同作用对低密度不饱和聚酯树脂制备的影响外文翻译资料
- 利用钢渣和草酸氢钾制备新型化学键合陶瓷外文翻译资料
- A位空位型钛酸铋钠基弛豫铁电体 具有超高的能量密度和更高的放电效率外文翻译资料
- 用热分析方法测定含氯化钠和氯化钾的油 井水泥浆体的水化产物外文翻译资料
- 将垂直排列的石墨烯片多孔膜用于高效太阳能热净水外文翻译资料

