
英语原文共 7 页,剩余内容已隐藏,支付完成后下载完整资料
振动光谱研究使用从头算和密度泛函理论分析5-氨基邻甲酚的结构
安纳马莱大学物理系(工程)系,Annamalai Nagar 608 002,泰米尔纳德邦,印度
Sri Aravindar艺术和科学学院物理系,Akasampet,Vanur District 605 111,泰米尔纳德邦,印度
2006年3月11日收到; 2006年12月3日以修订形式收到; 于2006年12月16日接受
摘要
在固相中记录5-氨基邻甲酚(5AOC)的傅立叶变换拉曼和傅立叶变换红外光谱。用6-311G(d,p)基组和HF和密度泛函B3LYP方法计算平衡几何,谐振频率,红外强度和拉曼散射活性。规模理论波数显示与实验值非常吻合。标题化合物的热力学函数也在HF/6-311G(d,p)和B3LYP/6-311G(d,p)理论水平下进行。报道了5-氨基邻甲酚的红外和拉曼光谱的详细解释。已经构建了标题分子的FT-IR光谱的理论谱图。
关键词:FT-IR和FT-Raman光谱;从头算和DFT;5-氨基-邻-甲酚;振动分析
1.介绍
5-氨基邻甲酚在其已知代谢性质方面是重要的化合物。一些用于染发剂如酚类化合物(苯酚,百里酚,甲酚,对叔丁基苯酚,间苯二酚)的成分可引起过敏反应,如皮疹,皮肤炎症,刺激和皮炎或促进癌症[1,2]。
蛋白质的拉曼光谱不仅提供了主链结构的信息,还提供了侧链微环境的信息[3]。重要的氨基酸残基是用于蛋白质结构的拉曼光谱研究的色氨酸,酪氨酸和苯丙氨酸,因为这些芳族残基的侧链振动在可见光激发的拉曼光谱中是强烈的,并且进一步它们可以选择性地通过紫外线激发增强[3-13]。从特征芳香族侧链的知识,我们可以解释蛋白质拉曼光谱的振动模式。Harada和Takeuchi[11]研究了色氨酸侧链和酪氨酸侧链的振动模式。
这种化合物及其三种氘代衍生物在液态下的拉曼光谱和液体溶液,蒸气和固态下的红外光谱已由Jakobsen报道[14]。Davy-Dova等人[7]计算了平面外振动的正常频率并分配了一些红外和拉曼频带。Green等人[10]提出了非氘代对甲酚基本频率的完整分配。我们还报告了用Wilson开发的经典方法对6-氨基间甲酚的正常坐标分析,以支持振动分析[15]。
文献调查表明,据我们所知,迄今为止还没有报道完整的拉曼和红外光谱以及5-氨基邻甲酚的力场。因此,本研究旨在完全研究该分子的振动光谱,并识别波数精度较高的各种正常模式。从头算HF和密度泛函理论(DFT)计算已经完成,以支持我们的波数分配。由Rastogi等人报道的具体比例因子[16]从苯分子推导出来已被用于预测5-氨基邻甲酚环模式的数量。
2.实验
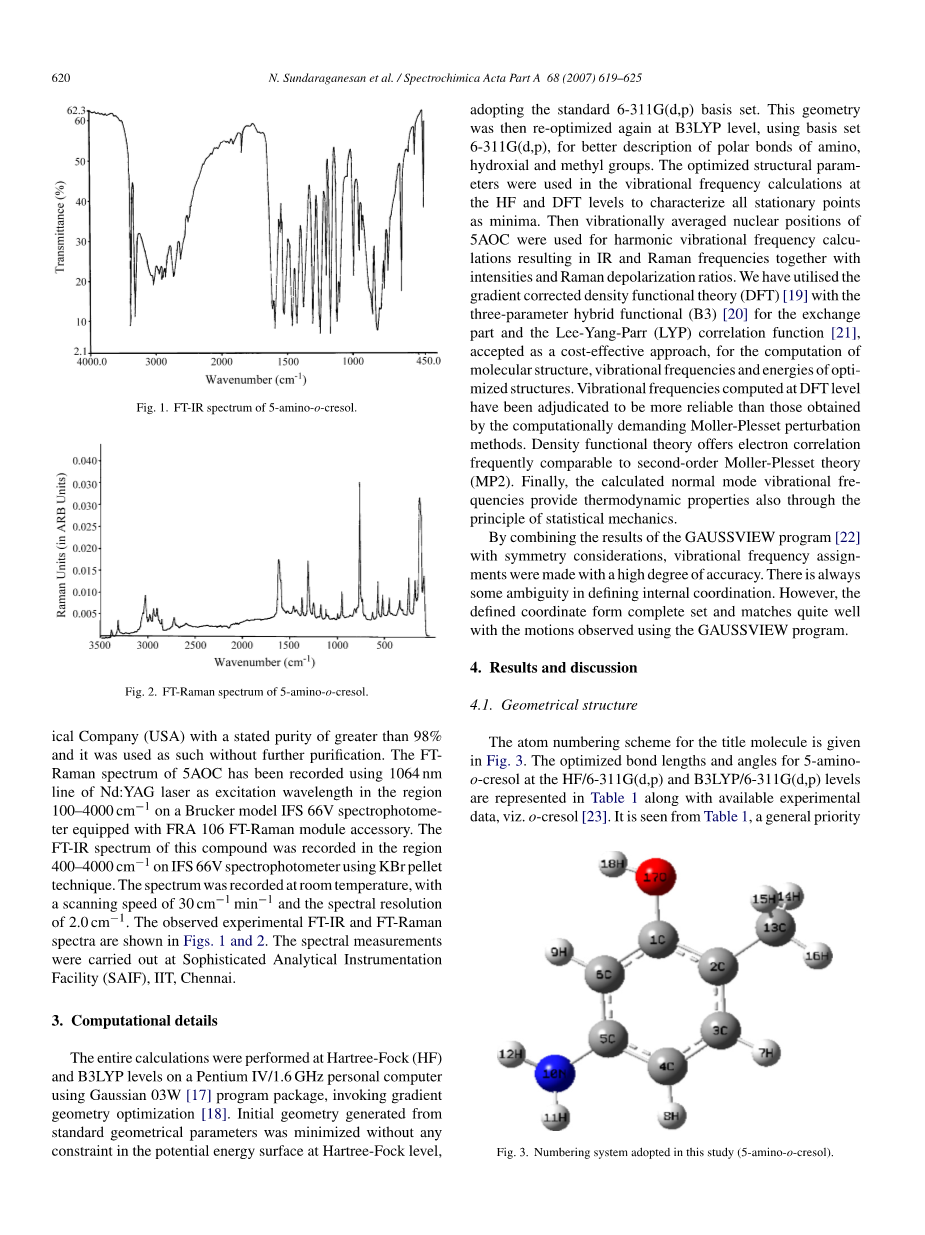
固体形式的化合物5-氨基-邻甲酚(5-氨基-2-甲基苯酚)购自Sigma-AldrichChemicalCompany(USA),纯度大于98%,并且其原样使用进一步纯化。使用1064nm的Nd:YAG激光线作为100-4000cm-1区域的激发波长,在配备有FRA106FT-Raman模块附件的Brucker型IFS66V分光光度计上记录5AOC的FTRaman光谱。使用KBr颗粒技术在IFS66V分光光度计上在400-4000cm-1的区域记录该化合物的FT-IR光谱。在室温下记录光谱,扫描速度为30cm-1min-1,光谱分辨率为2.0cm-1。观察到的实验FT-IR和FT-拉曼光谱显示在图1和2中。光谱测量是在精密分析仪器设备(SAIF),IIT,钦奈进行的。
图1. 5-氨基邻甲酚的FT-IR光谱 波数(cm-1)-透射比(%)
图2. 5-氨基邻甲酚的FT-拉曼光谱 波数(cm-1)-拉曼单位(在ARB单位)
3.计算细节
整个计算是在PentiumIV/1.6GHz个人计算机上使用Gaussian03W[17]程序包在Hartree-Fock(HF)和B3LYP水平上进行的,调用梯度几何优化[18]。采用标准的6-311G(d,p)基组,在Hartree-Fock水平的势能面上没有任何限制地将由标准几何参数产生的初始几何结构最小化。然后使用基组6-311G(d,p)在B3LYP水平上再次重新优化该几何形状,以更好地描述氨基,羟基和甲基的极性键。在HF和DFT水平的振动频率计算中使用优化的结构参数来将所有静止点表征为最小值。然后将振动平均的5AOC核位置用于谐振频率计算,得到IR和拉曼频率以及强度和拉曼去极化比。我们利用梯度修正密度泛函理论(DFT)[19]和交换部分的三参数混合功能(B3)[20]和Lee-Yang-Parr(LYP)相关函数[21]一种经济有效的方法,用于计算优化结构的分子结构,振动频率和能量。振动频率计算出来的水平已经被判定为比通过计算要求的莫勒-普莱塞斯扰动方法获得的更可靠。密度泛函理论提供的电子相关性经常与二阶Moller-Plesset理论(MP2)相当。最后,计算的正常模式振动频率也通过统计力学原理提供热力学性质。
通过将GAUSSVIEW程序[22]的结果与对称性考虑相结合,振动频率分配具有高度的准确性。定义内部协调总是有些含糊不清。然而,所定义的坐标形式是完整的,并与使用GAUSSVIEW程序观察到的运动相匹配。
4.结果和讨论
图3. 本研究采用的编号系统(5-氨基邻甲酚)
4.1.几何结构
在图3中给出了标题分子的原子编号方案。在HF/6-311G(d,p)和B3LYP/6-311G(d,p)水平下,优化的5-氨基甲酚的键长和角度分别为如表1所示,以及可用的实验数据,即,邻甲酚[23]。从表1可以看出,从参考文献[23]中重现实验键长的一般优先级,在HF和DFT-B3LYP水平中不存在。然而,与HF水平相比,用DFT-B3LYP水平计算的所有键长和键角与可用实验结果显示出极好的一致性。
键长的减少在B3LYP水平与HF水平相比更显着。C-CH3键的长度在两个水平都略高估,而C-O键的长度在HF水平被低估,在B3LYP水平略高估。
在苯胺中,氮原子离开环面大约2-3°(倾斜角),并且由氨基和平面定义的平面之间的角度为38plusmn;4°[24]。有关氨基非平面度的实验数据在文献中没有提供对甲基苯胺[25]。在5-氨基邻甲酚中,HF和DFT-B3LYP方法计算的氨基和环之间的倾角和角度与苯胺和对甲基苯胺的实验数据非常相似[25]。
表格1 在5-氨基邻甲酚中的几何参数优化,键长(A°)和角度(◦)
4.2.振动分配
根据理论计算,5-氨基邻甲酚具有Cs点对称的平面结构。该分子具有18个原子和48个正常的基本振动模式,跨越不可约表示:33Aˊ 15A'。在FT-IR和FTRaman中,所有48种基本振动都是有效的。
在表3中收集了使用三重分裂基价格集合以及扩散和极化函数6-311G(d,p)在B3LYP水平下计算的5AOC的谐振频率。观察到的FT-IR和FT-拉曼频率在表2中列出了各种振动模式。在B3LYP中计算的频率与实验值(表2)的比较表明,由于忽略了实际系统中的非谐性,所以计算的振动模式被高估。将密度泛函理论中的电子相关性包含在一定范围内使得频率值与HF频率数据相比更小。计算出的谐波振动减少,尽管基准设置敏感是边际的,如在使用6-311G(d,p)的DFT值中观察到的那样。任何不能抵抗计算水平的方式,都习惯于缩小计算出的谐波频率,以便改善与实验的一致性。在我们的研究中,我们采用了两个不同的比例因子(即0.9089至800cm-1和0.8992超过800cm-1),这些值输入表3的#39;a#39;列。#39;b#39;列包含值按照表3的脚注所示按不同的比例缩小。使用6-311G(d,p)在B3LYP水平上5AOC的棒光谱已经显示在图4中。
4.3.OH振动
OH基团振动可能对环境最为敏感,因此它们显示了氢键物质谱图的变化。在未取代苯酚的情况下,已经表明在气相中OH伸缩振动的频率是3657cm-1[26]。在我们的例子中,在3387cm-1处的FT-IR光谱中的强带和在3390cm-1处的FT-拉曼中的弱带被指定为OH伸缩振动。将这些谱带与文献资料进行对比可以预测,〜270cm-1的负偏差可能是由于强分子内氢键的存在。但是,B3LYP/6-311G(d,p)水平的计算值显示为3682cm-1(模式48)。
苯酚中的OH面内弯曲振动通常位于1150-1250cm-1区域,并且不像拉伸和平面外变形频率那样受氢键影响较大。在3-氨基苯酚中发现这种振动在1178cm-1[27]。在1173cm-1处FT-IR频率非常强,在1180cm-1处FT-Raman频率很弱,这归因于这种振动。B3LYP/6-311G(d,p)方法在1178cm-1处的理论计算值与实验观察结果完全一致。
苯酚OH的面外变形振动位于游离OH290-320cm-1区域和OH517-710cm-1区域[28]。在分子内和分子内结合中,频率都比游离OH中的更高。频率随着氢键强度的增加而增加,因为扭曲O-H键外面所需的能量较大[29]。在3-氨基苯酚的拉曼光谱[27]中,在350cm-1的谱带被分配为OH面外变形。振动在360cm-1时的计算值(见表3模式8)与文献值有很好的一致性。FT-IR和FT-Raman均未发现360cm-1的计算值。
表2 实验FT-IR和FT-拉曼频率和作业的5-氨基甲酚(cm-1)
图4. 在每个考虑的计算水平归一化IR强度中校正频率cm-1的比较
4.4.CO振动
5-氨基邻甲酚中的CO伸缩振动在模式编号中有主要贡献。28,B3LYP/6-311G(d,p)的预测频率为1212cm-1(表3)。这与在FT-IR光谱中1205cm-1处非常强的实验频率非常一致。理论频率为237cm-1的C-O面外弯曲振动模式与实验FT-拉曼值偏差约104cm-1。这可能是由于CO振动与CCC外面弯曲振动混合造成的。以上结论与文献值相符[30]。
4.5.C-H振动
由于5-氨基邻甲酚是三取代的芳族体系,因此它具有两个相邻的和一个孤立的C-H部分。预期的三个C-H伸缩振动对应于43,44和45.比例振动,43,44和45(表3)的列“b”对应于C3-H,C4-H和C6-H单位。分配给3047-3077cm-1区域芳香C-H伸缩的振动43-45与实验分配3012-3048cm-1一致[32]。即使发现被-NH2摇摆和OH平面内弯曲污染,区域1081-1288cm-1(模式24-26)中指定的C-H面内弯曲振动处于文献中的范围内[33,34],而实验观察在1130-1259cm-1。对于C-H平面外弯曲的计算频率755-912cm-1(模式号18-20)落在812-959cm-1的FT-IR/FT-拉曼值中。
表3 在B3LYP / 6-311G(d,p)处获得5-氨基邻甲酚的振动波数
4.6.C-NH2振动
在3417-3508cm-1(模式编号46和47)范围内的缩放的-NH2对称和不对称伸展与FT-IR中的〜3323cm-1和FT-Raman中的3326cm-1的实验值一致。在FT-IR和FT-Raman中,〜3508cm-1处计算出的不对称振动都不存在。计算出的1605cm-1处的-NH2剪切振动与预期的特征值1600cm-1非常吻合[35,36]。这与FT-Raman值为1624cm-1(模式38)的记录介质强带也有很好的一致性。NH2剪切模式也有助于在1618cm-1(模式号39)下的C-C拉伸模式。计算出对应于C-NH2部分的1317cm-1处的中频带FT-拉曼值为1304cm-1(模式号30)。在280和592cm-1处的C-NH2面外和面内弯曲振动分别与实验数据中的分配也很吻合。在641cm-1(模式号14)计算的NH2摆动与在635cm-1处的FT-拉曼值完全匹配。在FT-IR和FT-Raman光谱中,NH2扭曲振动计算为1081cm-1(模式编号24)。
4.7.C-C振动
预测在1579cm-1处C-C芳香族拉伸被称为半圆形拉伸,与在1560cm-1的FT-IR值的实验观察结果非常一致。在755cm-1(17号模式)下的环形呼吸模式分别与767cm-1处的非常强烈的拉曼谱带和FT-IR中759cm-1处非常弱的谱带令人满意地吻合[37]。已经发现理论计算的C-C-C面内弯曲和面外弯曲模式与记录的光谱值一致。
4.8.甲基振动
所考虑的标题分子5-氨基-邻甲酚在环的第二位具有一个CH3基团。对于CH3组频率的分配,人们可以预期9个基本原理可以相关联,即,CH3(CH3对称拉伸)中的对称拉伸和不对称拉伸(即面内氢拉伸模式);对称(CH3sym。变形)和不对称(CH3asym。变形)变形模式;面内摇摆(CH3ipr),面外摇摆(CH3opr)和扭曲(tCH3)弯曲模式。频率低于芳环(3000cm-1)的CH拉伸。对于CH3化合物,模式nu;s出现在2860-2935cm-1范围内,其中nu;as模式出现在2925-2985cm-1区域内。在2943/2948和2903/2911cm-1处的FT-IR/FT-
全文共6396字,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[11855],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 播撒生物炭促进鸟粪石形成,但加速重金属积累外文翻译资料
- 钢铁工业余热有机朗肯发电的能量及炯分析外文翻译资料
- 深度共晶溶剂微波辅助处理木质素-碳水化合物复合 物的高效裂解及超快提取木质素低聚物外文翻译资料
- 功能化杯状芳烃离子团族[4]的合成、晶体结构及竞争结合性能外文翻译资料
- 面向高能量密度柔性超级电容器的无纺布用黑磷杂化微纤维的微流控纺丝结构外文翻译资料
- 活性炭对水溶液中氨的吸附外文翻译资料
- 制备可控海胆状NiCo2S4微球协同硫掺杂石墨烯作为高性能 二次锌空气电池的双功能催化剂外文翻译资料
- 钛酸盐材料对重金属离子的吸附外文翻译资料
- CO2敏感催化剂的合成与表征温度响应催化聚离子液体微凝胶外文翻译资料
- 温度响应微凝胶薄膜在湿环境中作为可逆二氧化碳吸收剂外文翻译资料

