
英语原文共 16 页,剩余内容已隐藏,支付完成后下载完整资料
多面体齐聚硅氧烷(POSS)聚合物复合材料的分子动力学模拟
摘要:采用分子动力学(MD)模拟方法,研究了一种新型的含多面体低聚硅氧烷(POSS)的牙科纳米复合树脂的物理性能,并提出了一种交联聚合物模型的改进方法。通过宽X射线散射(WXRS)和体温特性验证了该技术的优势。良好的一致性证明了模型和力场的准确性,预测了各种POSS树脂的力学性能和体积收缩率,并与现有的实验数据进行了比较,进一步分析了POSS对牙科复合树脂的影响。改进后的方法可用于新型复杂结构交联牙科材料的进一步研究。
- 引言
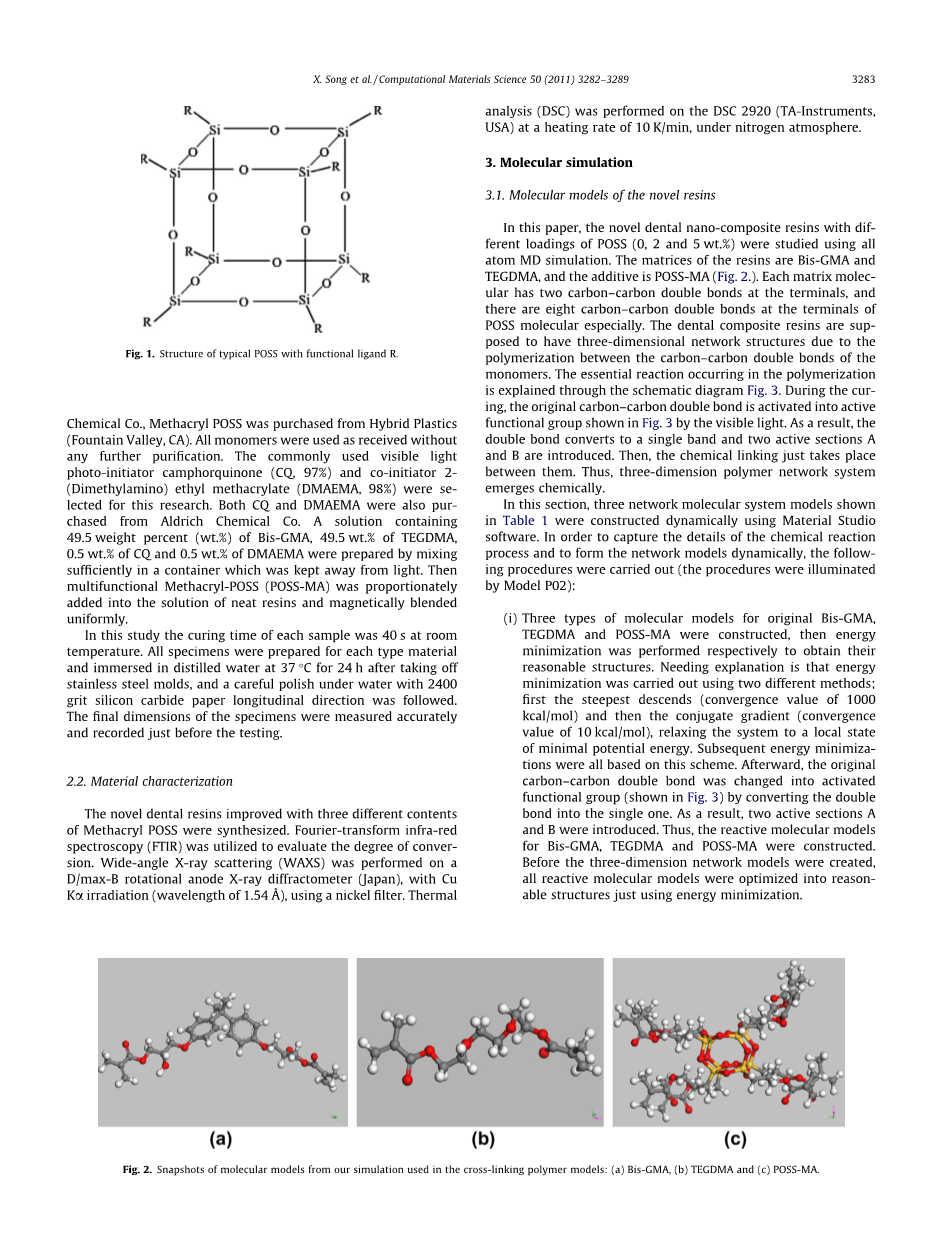
近几十年来,聚合物纳米复合材料由于其结构的可裁剪性和纳米级的优异性能,以及对标准复合材料具有更高的化学保真度而获得了广泛的关注。尤其是基于POSS的杂化有机-无机纳米复合材料在聚合物基体中的应用受到了广泛的关注。POSS具有紧凑的混合结构(如图1),具有由硅和氧原子(Sio1.5)n组成的无机核,其中n=8、10、12,外被非反应性或反应性有机配体所包围。显然,活性有机配体R的这种化学性质在控制杂化材料的形貌和功能性质方面起着重要作用。近年来它被广泛应用于牙科复合树脂中,以提高牙科树脂材料的耐磨性、力学性能、生物相容性和加工性能。
图1 具有功能配体R的典型POSS的结构
然而,目前国内外对POSS或其它牙科复合材料树脂的研究大多局限于实验技术上,由于其结构过于复杂,因此很少有研究通过MD模拟来解释其增强机理。以往的MD模拟主要集中在线性均质聚合物或共聚物上,而对网络聚合物的研究较少。一些网络聚合物是在MD模拟的基础上进行了研究,其中网络结构直接被线性链或简单的网段所取代。因为缺乏网络的细节,这种简化不可避免地降低了计算精度。近年来人们提出了几种方法,通过分子模拟并结合化学反应路径,对网络聚合物模型进行了化学构建,从而得到了较为合理的结果。
本文的目的是研究一种新型牙科纳米复合树脂与多功能多面体齐聚硅氧烷(POSS)结合的物理性能。在前人实验研究的基础上,提出了一种改进的牙科纳米复合树脂交联聚合物模型。随后,通过宽X射线散射(WXRS)和玻璃化转变温度Tg对模型的质量进行了MD模拟验证,模拟结果与相应的实验数据吻合较好。最后,用分子观方法对三种不同树脂模型的力学性能和体积收缩率进行了计算和分析。并将其结果与现有的实验数据进行了比较。
- 实验
2.1材料制备技术
双酚A甘油二甲基丙烯酸酯(Bis-GMA)和三乙二醇二甲基丙烯酸酯(TEGDMA)是从奥尔德里奇化学公司购买的,POSS是从混合塑料(喷泉谷,CA)购买。所有单体均按接收到的方式使用,不作任何进一步的纯化。本研究选用了常用的可见光光引发剂樟脑醌(CQ,97%)和共引发剂2-(二甲氨基)甲基丙烯酸乙酯(DMAEMA,98%)。CQ和DMAEMA也是从奥尔德里奇化学公司购买的。将含质量分数(wt.%)为49.5%Bis-GMA 、49.5 wt.% TEGDMA、 0.5 wt.%CQ和0.5 wt.%DMAEMA 的溶液充分混合在一个远离光线的容器中,然后在纯树脂溶液中按比例加入多功能甲基-POSS(POSS-MA),并进行磁均匀混合。
在本研究中,每个样品在室温下的固化时间为40 s。所有试样均为一种材料制备,取出不锈钢模具后,浸泡于37℃的蒸馏水中24 h,然后在水中用2400砂砾碳化硅纸仔细纵向抛光。在测试前,我们准确地测量和记录了试样的最终尺寸。
2.2材料特性的描述
合成三种不同含量的甲酰基新型牙科树脂,利用傅里叶变换红外光谱(FTIR)对其转化率进行了评价。在日本D/max-B旋转阳极X射线衍射仪上进行了宽角度X射线散射(WAXS),并用镍滤光片进行Cu Kalpha;辐照(波长1.54 Aring;)。关于DSC的最新分析是在氮气的环境中,以10 K/min的加热速率在美国TA仪器DSC 2920上进行的分析。
- 分子模拟
3.1新型树脂的分子模型
本文采用全原子MD的模拟方法,研究了不同POSS(0、2和5 wt.%)负载量的新型牙科纳米复合树脂。树脂的基体为Bis-GMA和TEGDMA,添加剂为POSS-MA,如图2所示。每个基质分子在末端有两个碳-碳双键,尤其是在POSS分子的末端有8个碳-碳双键。牙科复合树脂由于单体碳-碳双键之间的聚合,被认为具有三维网络结构。示意图3解释了聚合过程中发生的基本反应。固化过程中,在可见光下原碳-碳双键被活化成活性官能团,如图3所示。因此双键转换为单波段和两个活动部分A和B,然后在它们之间就会发生化学连接。由此,三维聚合物网络体系应运而生。
- (b) (c)
图2 在交联聚合物模型中使用的模拟分子模型的快照:(a) Bis-GMA,(b) TEGDMA和(c) POSS-MA。
最初 触动 网格
图3 聚合反应中的本质反应(红线呈现新的共价键)
在本节中,使用Material Studio软件动态地构建了表1所示的三个网络分子系统模型。为了捕捉化学反应过程的细节并动态地形成网络模型,执行了以下步骤(P02模型说明了这些程序):
表1 三种不同的交联模型在模拟和实验中的特性
- 建立了Bis-GMA、TEGDMA和POSS-MA三种分子模型,并分别进行能量最小化,得到了它们合理的结构。需要解释的是,能量最小化采用了两种不同的方法:首先是最陡的下降(收敛值为1000 kcal/mol),然后是共轭梯度(收敛值为10 kcal/mol),使系统处于最小势能的局部状态。随后的能量最小化都是基于这个方案。随后将原碳-碳双键转变为活性官能团(如图3),将双键转化为单键。因此引入了两个活动部分A和B,从而建立了Bis-GMA、TEGDMA和POSS-MA的反应分子模型。在建立三维网络模型之前,所有的反应分子模型都被优化为合理的结构。
- 其次,利用 Material Studio软件中的非晶胞模块,将64个Bis-GMA活性分子模型、100个TEGDMA活性分子模型和1个POSS-MA活性分子模型封装在一个密度为1.2121 g/cm3的立方晶格中。为了消除表面效应,引入了三维周期边界条件,然后建立新型牙科纳米复合树脂模型的初始物理混合模型。
- 第三,采取了50,000次能量最小化的措施来放松单个细胞系统。在此基础上,对松散的物理混合物体系进行评价,以识别近距离的活化段。首先选择一个活化段的A或B的碳,然后再对附近活化段的碳进行另一次搜索。在反应截止距离范围内,只有最接近的两个活化段的碳才能发生反应,这意味着它们之间将形成共价键。特别是,本文选择从3到10 Aring;的截止距离是因为这个选择可以避免在同一活性官能团的两个附近的碳发生反应。另外根据实际情况,截止距离越大越好,并且在反应过程中应避免形成四元环和五元环。虽然对真实的交联系统进行了尽可能的模拟,但也允许存在链纠缠等物理缺陷。
- 最后,对改性的分子模型体系进行了重新验证,以评价聚合过程中双键的转化。如果双键转化率小于FTIR的实验结果,则最大的截止距离扩大并再次执行第三步。另一方面,如果转化率大于红外光谱的实验结果,则最大的截止距离减小,并再次进行第三步。直到双键转换下降到允许的范围内,程序结束并且系统中的所有非氢原子都被氢原子饱和。选择最终的物理和化学混合物模型系统进行进一步的研究。
所有步骤都被编码为Perl脚本,并在Accelrys的Material Studio软件中运行。
最后,在表1和表2中建立了三个不同POSS负载量(0、2.02和5.02 wt.%)的网络分子系统模型。很明显,模拟中网络聚合物POSS的负载与实验值接近,各种网络聚合物的密度在一定的误差范围内也与实验结果相吻合。
表2 三种不同交联模型的聚合特性
注:聚合转化率由双键的转化率,即转化率=(固化双键数)/(原总双键数)来评价。
3.2分子模拟方案
所有的模拟都是基于COMPASS(原子模拟研究的凝聚相优化分子势)力场进行的。COMPASS力场是第一个用凝聚相性质参数化和验证的力场,它在许多POSS纳米复合材料上得到了有效的应用。在COMPASS力场中,总能量表达式有键相互作用和非键相互作用两个主要项。后一种相互作用(范德华和静电能)在总能量表达式中是非常重要的术语。由于能量项取决于系统中原子的数目,因此太远的原子之间的相互作用应该被忽略。整个研究的截止点为9.5Aring;,而基于原子的方法被选择为范德华尔能量。然而,这种方法可能会导致静电相互作用的能量不连续,特别是对于电荷较大的系统。因此,本文选择了精度较高的Ewald法作为静电能量的最佳计算方法。用周期边界条件模拟材料,Noseacute;算法更稳定,能够保持系统的规则性。因此,它被选择用来控制温度。Parrinello算法使模拟单元的对称性和尺寸都发生变化,因此选择它来控制压力。用Verlet积分器的速度形式求解运动方程,时间步长为0.5fs。在进行MD模拟之前,对单元系统进行了50,000次能量最小化。随后,对选定的网络系统模型,在恒定粒子数、压力和温度(NPT)条件下进行了MD模拟。P00和P05网络系统模型的快照如图4所示。
- (b)
图4 模拟快照(a)网络聚合物模型P00和(b)聚合物模型P05
注:在P05模型中,POSS呈橙色椭球状。
4.结果与讨论
4.1宽X射线散射剖面
计算了所选形状完全平衡的宽X射线散射剖面。图5对仿真结果与实验结果进行了比较,模拟结果与实验结果在峰位和强度上吻合较好。可以得出结论研究中建立的网络系统聚合物模型、力场模型和分子动力学模型是有效的。另外,不同POSS负载量的网络聚合物在WXRS上表现出相似的模拟和实验趋势,只是WXRS的强度略有不同。这些现象表明,不同POSS负载量的新型网络聚合物的结构是相似的。对于所有聚合物树脂,WXRS图谱显示2theta;=18.50ordm;处有一个宽的非晶晕,对应于4.43Aring;的有效布拉格距离,这一现象证实了网络聚合物树脂是无定形的。在2theta;=41.18ordm;的条件下,实验结果中还存在一个较小的非晶晕,而在相同的位置上模拟的光晕并不明显。
图5 模拟和实验WAXS结果: (a) P00, (b) P02和 (c) P05
4.2玻璃化转变温度
本研究采用NPT MD模拟方法,确定了体积与温度的关系,并对Tg进行了预测,在体积温度图上斜率变化的温度表示聚合物的Tg。为了确保模型完全平衡,在网络系统上进行了MD模拟,首先将温度从600 k降到50 k,然后从50 k升到600 k,最后以每50 k为间隔从600 k冷却到50 k。只选择最后的冷却过程来分析网络聚合物模型的Tg。此外,在每个等温温度下MD模拟超过250 ps,并且用10种以上不同构型的平均体积来确定体积-温度图。在图6中给出了不同载荷下POSS模型的体积-温度图。
图6 体积-温度曲线(a) P00, (b) P02和 (c) P05
在每一种情况下,斜率的转折都是明显的,这表明网络聚合物模型的尺寸足够大,足以评估玻璃化转变行为。
用体积-温度图模拟玻璃化转变温度,并与DSC的实验结果进行了比较(如图7所示)。模拟结果与实验结果吻合较好,两者均在50~60℃范围内。这些结果进一步验证了分子模拟方案和从模拟中计算Tg的方法。
图7 POSS加载下玻璃化转变温度的模拟与实验
另外,结果还表明POSS的加入对玻璃化转变温度的影响不大。这种结果很容易理解,因为改进后的牙科树脂和原树脂具有相似的网络结构。模拟结果略高于实验结果,类似的现象在许多文献中都有报道。一般来说,聚合物的玻璃化转变发生在一定的温度范围内。冷却速率对玻璃转变温度的测量有影响,模拟中重复单位的数目也会使玻璃转变温度发生变化。在MD模拟中,冷却速率(1011 k/s)明显高于实验(10 k/min)。这些因素使模拟结果在一定程度上不同于实验结果。因此,本节的结
全文共6159字,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[9070],资料为PDF文档或Word文档,PDF文档可免费转换为Word

