
英语原文共 4 页,剩余内容已隐藏,支付完成后下载完整资料
2,4,5-三取代氧化唑的制备
摘要:以碘介导的烯胺酮衍生物有氧氧化环化为基础,开发了一种新的三取代恶唑的方法。这种无过渡金属的方法非常高效,涉及了在温和条件下去除四个氢原子的过程。
近年来,恶唑引起了人们的广泛关注1,因为来源于海洋无脊椎动物和微生物的噁唑类化合物显现出多样化的生物活性。2另外,噁唑基团在生物活性天然产物和药物的合成中广泛使用。3许多有前景的抗菌,3e抗糖尿病,3h和抗炎3i确定了可以合成有活性的三取代恶唑(方案1a)。因此,为实现完全取代的恶唑环系统的合成做出了巨大的努力。
i
方案1。 多取代氧化唑的合成方法
迄今为止,已经通过酰基前体的环化制备了一系列高度功能化的恶唑。 尤其是由布朗斯台德或路易斯酸(称为罗宾逊minus;加布里埃尔缩合反应)4促进的alpha;-环酰胺酮、酯类或酰胺的环脱水作用是合成恶唑衍生物的经典策略。 过渡金属催化5或者碘6介导的酰胺环化反应也得到了广泛的应用。例如2012年,Jiao和他的同事开发了通过铜介导的氧化脱氢环反应从醛和胺中合成2,5-二取代恶唑(方案1b)。7在2015年,高和同事报告了一种通过I 2催化Cminus;O键的形成/beta;-酰基氨基酮(方案1c) 合成2,4,5-三取代恶唑和恶唑啉。8然而,上述大多数策略有一些缺点,如需要使用强质子酸或化学计量的氧化剂甚至是过渡金属催化剂。基于这些挑战,因此我们仍需研究怎样在温和的反应条件下合成高功能化恶唑。 因此我们研究如何将氨基转化为含氮杂环。9以烯胺酮为原料,采用碘作为催化剂,通过氧化脱氢环法合成2,4,5-三取代恶唑衍生物。反应过程是在无过渡金属条件下,四个氢原子被有效地去除。 此外,分子氧作为氧化剂有利于合成含氧的杂环。
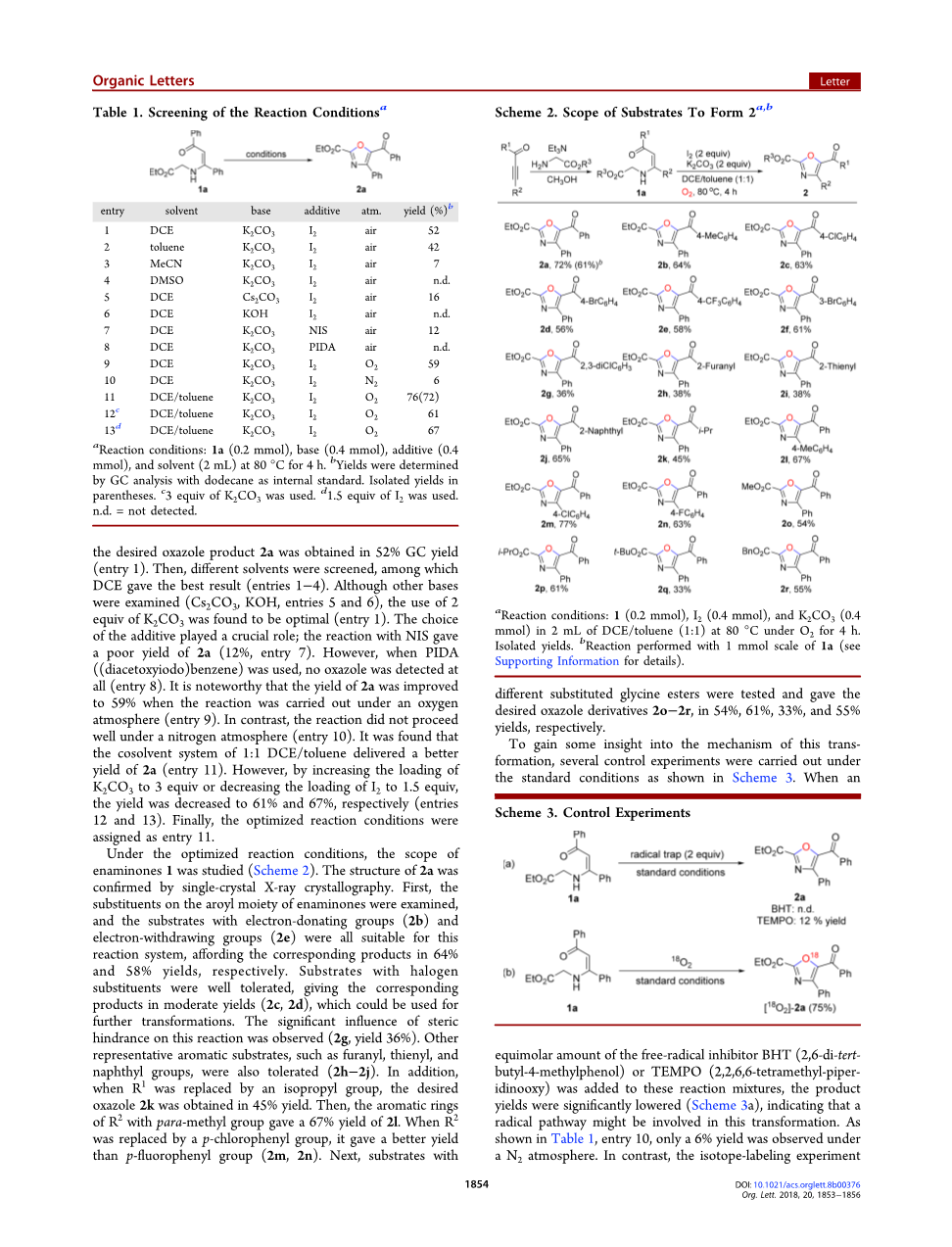
以N-(3-氧-1,3-二苯基丙-1-烯-1-基)-甘氨酸乙酯1a为底物,碘为添加剂,K2CO3为碱性溶液,二氯乙烷 (DCE)为溶剂,在80℃下对反应条件进行了初步研究 (表1)。令我们高兴的是,所需的恶唑产物2a的GC收率为52%(条目1)。然后,筛选了不同的溶剂,其中DCE效果最佳(条目1-4)。尽管检查了其他碱(Cs2 CO3,KOH,条目5和6),但发现使用2当量的K2CO3是最佳的(条目1)。添加剂的选择也至关重要。与NIS的反应收率很低,仅为2a(12%,条目7)。但是,当使用PIDA((二乙酰氧基碘)苯)时,根本没有检测到噁唑(条目8)。值得注意的是,当反应在氧气下进行时,2a的收率提高到59%(条目9)。但是该反应在氮气下进行得不好(条目10)。已发现1:1 DCE /甲苯的助溶剂体系可提供更高的2a收率(条目11)。但是,通过将K2CO3的用量增加到3当量或降低I2到1.5当量时,产率分别降低至61%和67%(条目12和13)。最后,将优化的反应条件指定为条目11。
|
入境 |
溶剂 |
基地 |
添加剂 |
atm |
产量(%)b |
|
1 |
DCE |
k2Co3 |
i2 |
air |
52 |
|
2 |
甲苯 |
k2Co3 |
i2 |
air |
42 |
|
3 |
MeCN |
k2Co3 |
i2 |
air |
7 |
|
4 |
DMSO |
k2Co3 |
i2 |
air |
n.d。 |
|
5 |
DCE |
Cs2Co3 |
i2 |
air |
16 |
|
6 |
DCE |
KOH |
i2 |
air |
n.d。 |
|
7 |
DCE |
k2Co3 |
NIS |
air |
12 |
|
8 |
DCE |
k2Co3 |
PIDA |
air |
n.d。 |
|
9 |
DCE |
k2Co3 |
i2 |
o2 |
59 |
|
10 |
DCE |
k2Co3 |
i2 |
n2 |
6 |
|
11 |
DCE/甲苯 |
k2Co3 |
i2 |
o2 |
76(72) |
|
12c |
DCE/甲苯 |
k2Co3 |
i2 |
o2 |
61 |
|
13d |
DCE/甲苯 |
k2Co3 |
i2 |
o2 |
67 |
a反应条件:1a(0.2mmol)、碱(0.4mmol)、添加剂(0.4mmol)和溶剂(2mL)在80°C下反应4小时。
b以十二烷为内标,GC分析测定产量。
C使用3当量的K2CO3。
d使用1.5当量的I 2。nd =未检测到。
在优化的反应条件下,对烯胺1的范围进行了研究(方案2)。 2a的结构通过单晶X射线晶体学确认。首先,检查烯胺酮的芳酰基部分上的取代基,具有供电子基团(2b)和吸电子基团(2e)的底物均适用于该反应体系,相应产物的产率分别为64%和58%产量。具有卤素取代基的底物具有良好的耐受性,以中等收率(2c,2d)得到相应的产物,可用于进一步转化。观察到位阻对该反应有显著影响(2g,收率36%)。其他代表性的芳香族底物,例如呋喃基,噻吩基和萘基,也可以耐受(2h–2j)。另外,当R1被异丙基取代时,以目标恶唑2k收率为45%。然后,具有对甲基的R 2的芳环的2I收率为67%。当R2被对氯苯基取代时,其收率比对氟苯基(2m,2n)更好。接下来,试验具有不同取代甘氨酸酯的底物,并分别以54%,61%,33%和55%的产率得到所需的恶唑衍生物2o-2r。
为了深入了解这种转化机制,如方案3所示,在标准条件下进行了一些对照实验。当将等摩尔量的自由基抑制剂BHT(2,6-二叔丁基-4-甲基苯酚)或2,2,6,6-四甲基哌啶子基氧基(TEMPO)添加到这些反应混合物中时,产量显着降低(方案3a),表明该转化过程可能涉及自由基途径。如表1的条目10所示,在N2条件下仅观察到6%的收率。与此相反,在标准条件下,用18ouml;2进行同位素标记实验得到[18O]标记的产物[18O]-2a,收率为75%。以上结果表明,恶唑产物的氧原子衍生自分子氧。
根据实验结果和文献报道,我们提出的该恶唑生成反应的机理在方案4展现。
方案4。 可能的反应机制
在碱性条件下,活跃的甘氨酸酯的Cminus;H被分子碘氧化得到自由基中间体3,11然后被双氧氧化成自由基中间体4。 然后,自由基4的异构化得到自由基5,其可能与I2反应生成中间体6。 然后,HOI可以从中间体6释放得到中间体7,遂得到噁唑产物2。
总之,我们开发了一种新的合成2,4,5-三取代恶唑的方法,该方法在碘介导下,烯丙酮衍生物与分子氧发生好氧脱氢环化反应。 这一过程涉及四个氢原子的去除和两个新的Cminus;O键的形成。 在这种反应中,利用分子氧作为恶唑的氧源,使该工艺环保、原子经济。 由于反应条件温和,官能团耐受性好,该方法在含恶唑类天然产物的合成中有广泛的应用前景。
■
- 作者信息
相应的作者
*电子邮件:glcheng@hqu.edu.cn.
ORCID
说明
提交人宣布没有相互竞争的经济利益。<!--
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[257496],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 下游产品是硫酸乙酰肝素2-0-磺基转移酶的有效抑制剂外文翻译资料
- 奥希替尼的耐药性不断发展的格局外文翻译资料
- 伪五山淫羊隆区域特异性鼠李糖基转移酶催化丙烯醇的3-阿霉素酰化 反应外文翻译资料
- 标记化合物和放射性药物杂志外文翻译资料
- 脂肪酶大规模分离水飞蓟宾非对映异构体外文翻译资料
- 钯催化萘胺与二芳基二硫化物和二硒醚的 C-H键裂解近选择性加成反应外文翻译资料
- 铜催化,定向基团辅助氟化芳烃和异芳烃碳氢键外文翻译资料
- Talin1通过局灶性粘附信号和失巢凋亡抵抗促进肿瘤侵袭和转移。外文翻译资料
- 杜氏肌营养不良综合征中的外显子跳跃外文翻译资料
- 来源于红树林内生菌稀有的细菌产caryolanes类代谢产物Bacaryolanes A−C外文翻译资料

