
英语原文共 11 页,剩余内容已隐藏,支付完成后下载完整资料
(1-x)Ba(Zr0.2Ti0.8)O3-x(Ba0.7Ca0.3)TiO3无铅压电陶瓷电荷密度及压电铁电性质的研究
摘要:实验分析的(1-x)Ba(Zr0.2Ti0.8)O3-x(Ba0.7Ca0.3)TiO3,(x=0.4,0.5和0.6)(简称(1-x)BCT-xBZT)无铅陶瓷是通过固态反应法所制备。X射线衍射实验结果表明,室温下在所有组分的陶瓷中只存在单一的钙钛矿相。XRD粉末精化分析的结果证实了没有其余的相,并且其计算得到的峰值强度与观察到的相吻合。电荷密度分布实验揭示了结构中化学键的性质。此外,结构中电子的定性和定量分布特征取决于单位晶胞中的掺杂原子。扫描电子显微镜图像表明粉体颗粒呈现均匀分布。通过利用能量色散X射线分光计,我们对样品的组成元素进行了分析。所有样品的光学带隙位于3.15到3.07电子伏特之间。在x=0.5的组分的准同型相界中,材料的压电常数最大,测得压电应变常数d33=276pC/N。
1.引言
压电材料,这种能实现电能和机械能的相互转化的材料,在传感器、驱动器、变压器及其他铁电器件领域已有广泛的应用。在过去的几十年里,锆钛酸铅陶瓷因具有良好的压电性能已得到广泛应用。但铅基PZT材料因成分中含有超过60%质量百分数的氧化铅,会造成各类环境问题及众多的疾病症状,如头痛、便秘、恶心、贫血、神经大脑和肾的损伤等。因此,为保护环境,铅基压电陶瓷已被无铅材料所取代。近些年来,无铅压电材料已引起科学界的广泛关注,大力发展无铅材料渐渐成为主题。与PZT材料相比,无铅材料的压电性能低得多,这是其长期存在的一个问题。
在各种无铅体系中,钛酸钡基系统具有优良的压电性能。刘等人首先报道了x[Ba(Zr0.2Ti0.8)O3]-(1-x)[(Ba0.7Ca0.3)TiO3]/[BZT-BCT]这种二元铁电准同型相界系统来取代传统的PZT基系统。最近有报道称,Ba(Zr0.2Ti0.8)O3-x(Ba0.7Ca0.3)TiO3无铅陶瓷在x=0.5时表现出压电应变常数接近620pC/N的高压电系数。组成为0.5 BZT-0.5BCT的材料在准同型相界处表现出优良的压电性能。此外,其与PZT及PMN-PT等铅基系统有着相似的相图。因此,BZT-BCT系统因其无铅特性而与铅基系统准同型相界相似且具有如文献报道所述的优良压电性能,是当前众多压电材料中的一个重要发展方向。
当前,科学界对(1-x)BZT-xBCT陶瓷的宏观和微观结构、光学、电介质、铁电和压电性能已有所研究。因此,我们利用最大熵值法,将研究重点放在了陶瓷的电子密度分布及键的性质上。
2.实验
实验分析的(1-x)Ba(Zr0.2Ti0.8)O3-x(Ba0.7Ca0.3)TiO3,(x=0.4,0.5和0.6) 无铅陶瓷通过固态反应法所制备。市购的BaCO3(99.99%)、CaCO3(99.95%)、TiO2(99.99%)、ZrO2(99.99%)粉体是实验的原材料。原材料按照化学计量比混合后球磨12小时。随后,混合粉末在1350摄氏度的氧化铝坩埚中煅烧2小时。煅烧后的粉末经过研磨与2%的作为黏合剂的聚乙烯醇(PVA)进行黏合并压制成直径12 mm、厚度1 mm的小球。然后,将小球在1450摄氏度的空气中以每分钟5摄氏度的升温速率烧结三小时。
我们利用粉体X射线衍射技术对烧结后的固溶体相结构进行分析。实验的XRD数据通过利用Cu的Kalpha;射线通过X射线衍射仪记录2theta;角在20°到120°的数据获得,其中扫描步长为0.02°。样品的表面形貌利用扫描电子显微镜进行分析,其在200-2000纳米波长范围内的紫外可见光谱图通过瓦里安卡里5000分光光度计记录。在颗粒样品经抛光两面涂上银浆后我们对其电性能进行了测试。样品的介电性能通过实现温度从40-200摄氏度的变化利用阻抗分析仪进行了测试分析。所有的极化过程均在30kV/cm的直流电场下40℃的硅油中极化20分钟完成的。通过利用辐射精密工作站在室温下我们完成了极化电场(P-E)磁滞回线的测量。压电常数d33通过d33测试仪完成测量。
3.结果与讨论
3.1结构分析
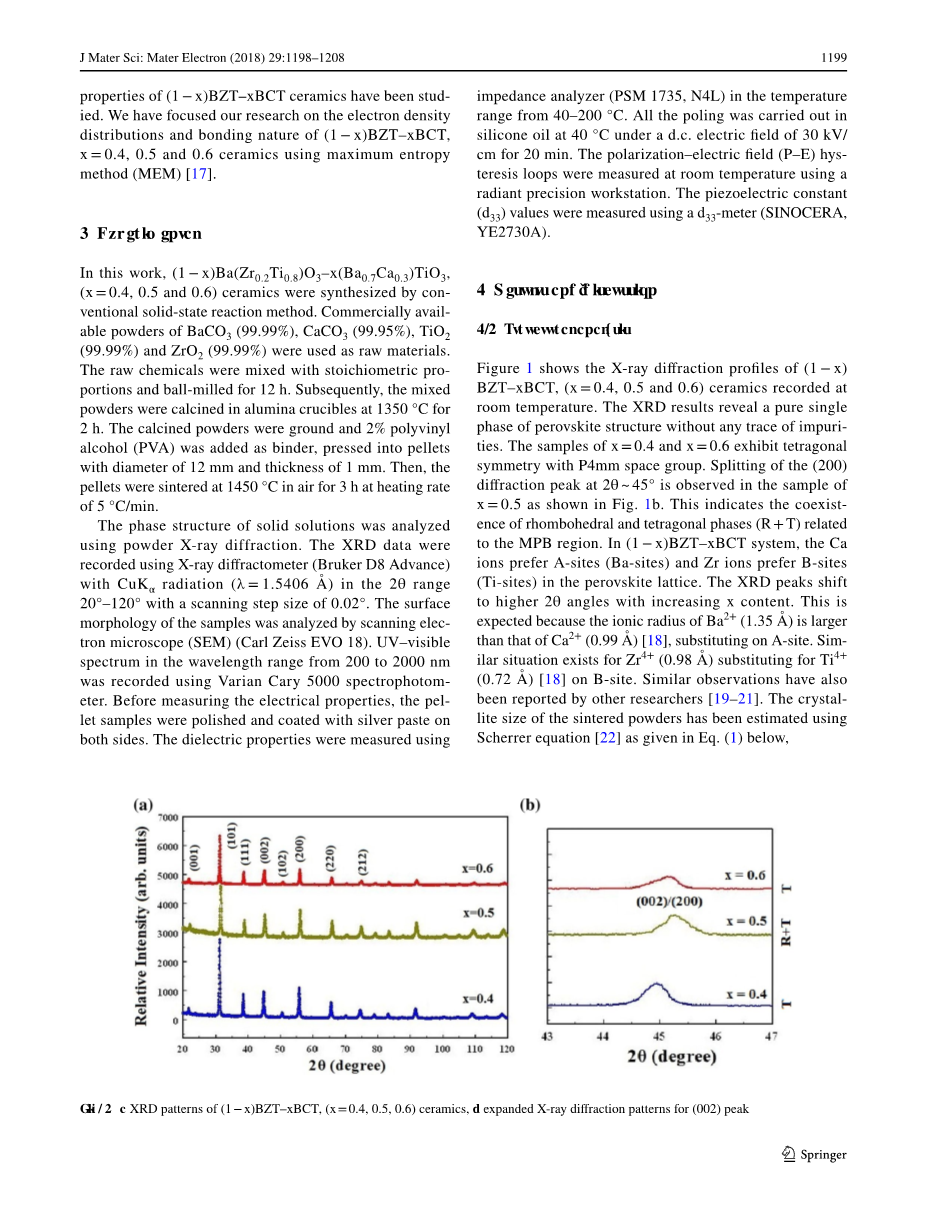
图1给出了(1-x)BZT-xBCT陶瓷在室温下的X射线衍射谱。XRD测试结果证实了室温下在所有组分的陶瓷中只存在单一的钙钛矿相。X=0.4和0.6的样品的P4mm空间点群表现出四方对称性,x=0.5的样品在2theta;为45°左右观察到如图1(b)所示的(200)衍射峰分裂,这表明四方相与菱方相的共存与准同型相界有关。在(1minus;x)BZT–XBCT体系中,钙钛矿晶格中的钙离子倾向于A位(Ba位),锆离子倾向于B位(Ti位)。当x含量增加时,x射线衍射峰向着高的2theta;角移动,这是由于钡离子的离子半径1.35埃大于A位点 取代的钙离子的离子半径0.99埃。离子半径0.98埃的锆离子在发生B位取代离子半径0.72埃的钛离子也有相似的情况,其他研究人员也已有所报道。我们可以通过谢瑞方程d=0.9lambda;/beta;costheta;来估算烧结粉末的微晶尺寸。其中,0.9为形状系数,lambda;为X射线波长(lambda;=1.54056埃),beta;是最大强度一半处以弧度表示的线展宽,theta;是布拉格角。(1-x)BZT-xBCT陶瓷(x=0.4、0.5、0.6)估计的平均晶粒尺寸分别约为27、18和25纳米。
图1
3.2里特菲尔德精化分析
本次测试使用Jana 2006软件对(1minus;x)BZT–XBCT(x=0.4、0.5和0.6)陶瓷样品的XRD实验数据进行了里特菲尔德精化。图2a-c显示了(1-x)BZT-xBCT(x=0.4、0.5和0.6)的四方结构模型各自的拟合数据和实验测试数据。在里特菲尔德分析中,通过将计算与观测得到的粉体衍射花样的差异最小化实现XRD数据的精化处理。从精化结果看来,钡离子和钙离子共享钙钛矿晶体结构中的A位点。相似地,锆离子和钛离子位于结构中的B位点。结构精化的结果表明,x=0.4和0.6的衍射峰可以用空间群p4mm表示为四方相。图2b显示了组分0.5BZT-0.5BCT精化分析在准同型相界处主要的四方相(P4mm)和弱的菱方相(R3m)的混合相。表1显示了使用里特菲尔德精化拟合后获得的晶格参数、可靠性指数及拟合优度。
图2
表1
3.3显微结构和能量色散X射线光谱分析
图3a–c显示了(1minus;x)BZT–xBCT(x=0.4、0.5和0.6)陶瓷的扫描电镜照片。x=0.4的组分的晶粒在扫描电镜下呈矩形。当x增加时,晶粒的形状随Ca2 浓度的变化而变化。可以发现,当Ca2 的浓度增加时,制备样品的平均粒径也相应增大。通过图4a-c的(1minus;x)BZT–xBCT(x=0.4、0.5和0.6)的能量分散X射线(EDS)光谱,我们可以得知固溶体的化学成分。表2列出了EDS光谱和EDS数据检测到的这些陶瓷样品中的所有元素的存在情况。EDS结果表明,观察到的原子和重量百分比与预期值吻合良好。
图3
图4
表2
3.4带隙测定
通过紫外-可见光谱分析估算出光学带隙能(Eg)。由伍德陶克方程(alpha;hv)2=A(hv-Eg)可知,光学带隙能Eg与吸收率和光子能量相关。公式中A是比例常数,alpha;是吸光度系数,h为普朗克常数, nu;是频率,Eg是光学带隙,n是一个与电子跃迁类型有关的常数。对于直接允许、间接允许、直接禁止、间接禁止的跃迁类型,其值分别为1/2、2、3/2和3。
通常情况下,我们通过绘制(alpha;hv)2与能量的关系图来进行带隙能的测定。通过将曲线的直线部分外推到光子能量轴,使得alpha;=0从而得出带隙能,如图5所示。表3中,(1-x)BZT-xBCT陶瓷(x=0.4、0.5、0.6)的光学带隙能值已给出。所有样品的带隙能值在3.15至3.07 eV之间,与报道值3.26 eV相比略小。
图5
3.5介电常数测量
(1-x)BZT-xBCT陶瓷(x=0.4、0.5、0.6)在10kHz下介电常数与温度的关系如图6所示。介电常数测量的温度变化范围在40-200摄氏度之间。居里温度(Tc)是铁电-顺电相变对应的温度。研究结果表明,随着x值的增加,居里温度Tc向着高温的方向发生移动。由于组分0.5BZT-0.5BCT在准同型相界处四方相和菱方相的混合结构,其具有最高的介电常数(ε ~ 3702)。菱方相和四方相的共存增加了该组分中极化取向的数量,这是其具有高介电常数的根本原因。表4给出了(1minus;x)BZT–XBCT(x=0.4、0.5和0.6)陶瓷在10 kHz下的介电常数值。
图6
表4
3.6铁电和压电测量
图7a-c显示了(1minus;x)BZT–XBCT(x=0.4、0.5和0.6)陶瓷在4.5 kV/mm外加电场下的室温极化与电场图。样品的最大极化值(PM)、残余极化值(PR)和矫顽力场(EC)如表4中所示。图7b显示了软p-e回路在x为0.5时取得最大极化值(Pm)15.12mu;c/cm2和矫顽力(Ec=3.62kv/cm)。0.5BZT-0.5BCT组分在室温下表现出最大的极化取向,这意味着一种与准同型相界类似的行为,正如XRD衍射图样所示。
我们测量了在40摄氏度下30kv/cm外电场作用下电极化样品的压电常数(d33)。由于组分0.5BZT-0.5BCT在准同型相界处四方相和菱方相的混合相结构的存在,混合相的存在为极化矢量的旋转提供了有利的条件,陶瓷的压电性能得到一定的改善,d33值达到了最大的276pC/N。由此可见,0.5BZT-0.5BCT陶瓷中存在的四方相和菱方相的准同型相界对其压电性能的改善具有重要的作用,这在固态反应法合成实验的文献中已有所报道。
图7
3.7电子密度分析
利用X射线衍射数据进行最大熵值法分析是推导原子间电荷密度分布以及相邻原子间相互作用和键合特性的一种简单方法。通过使用最大熵值法利用精化得到的XRD结构因子构建单位晶胞的电荷密度模型。最大熵法(MEM)是柯林斯(1982)提出的一种估算精确电荷密度的标准方法。电荷密度分布通过将每个单元晶胞考虑为64times;64times;64像素来进行分析的。均匀前置电子密度为固定值F000=/a03。其中,F000是单位晶胞的电子总数,a0是是单位晶胞的晶胞参数。我们通过利用Vesta软件实现了对单位晶胞内的预估电荷密度分布进行了可视化。图8a-c显示了几种组分的三维电荷密度分布状况。图9a显示了带有(0 0 1)晶面电荷密度分布的三维晶胞的电荷密度分布。图9b-d显示了(0 0 1)晶格平面均匀前置电子密度0-1e/埃3的二维等值线图,其中相邻两条等值线差值为0.04e/埃3。从图9b-d可以明显看出,Ba和O之间没有电荷重叠,这说明了Ba和O之间结合键的离子键性质。图10a显示了带有(0 0 2)晶面电荷密度分布的三维晶胞单元。图10b-d显示了(0 0 2)晶格平面均匀前置电子密度的二维等值线图,该等值线图表示了Ti和O离子之间的电荷密度分布状况。等值线图中Ti与O之间存在电荷重叠,这说明了Ti和O原子之间结合键的共价键性质。图11和图12分别表示了沿Ba–O和Ti–O键的一维电子密度分布状况。键长和中键电子密度值如表5中所示。
在钛酸盐钙钛矿(如PbTiO3和BaTiO3)结构中,Ti-O键的共价键性质是其能产生铁电极化的主要原因。表5给出了Ba-O键中键电子密度值从0.1689到0.1480 e/埃3的变化数据,这是导致Ba和O离子之间键性表现为离子键的原因。从表5中也可以知道,对于组成x=0.5的组分,Ti–O键中键电子密度值为0.9575 e/埃3。这进一步说明了Ti和O离子之间键性表现为共价键的原因。几种不同的组分在x=0.5时压电常数取得最大值d33~276pC/N,这正与 Pm~15.12mu;c/cm2的相对最大极化和Ec~3.62 kv/cm的相对较低矫顽力场相对应。可以得知,x=0.5的组分增强的压电和
全文共5523字,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[206],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 复杂热电材料外文翻译资料
- 以自蔓延高温烧结方法制备热电化合物以及燃烧合成的新标准外文翻译资料
- 氮掺杂分级多孔碳作为氧还原反应的高效电化学催化剂的研究外文翻译资料
- 孪晶诱导塑性高嫡合金的设计外文翻译资料
- 含铌先进Fe-Cr-Ni型奥氏体耐热钢富铜相的析出强化在超临界电厂的应用外文翻译资料
- 不同温度下直接能量沉积层状工具钢的弯曲强度外文翻译资料
- BiFeO3的光伏效应外文翻译资料
- 通过氢稳定的MgaPt研究核壳纳米结构Mg@Pt中快速“氢泵”的可视化外文翻译资料
- 一种铱核心环金属有机配体显著地提高了有机太阳能电池 的光伏性能外文翻译资料
- 钠离子电池的高性能阳极材料:三组分共组装法制备层次多孔碳外文翻译资料

