
英语原文共 4 页,剩余内容已隐藏,支付完成后下载完整资料
铁/ S8催化邻硝基芳烃与芳基甲基氯化物的氧化还原缩合:苯并咪唑和苯并噻唑的合成
甘海峰
摘要:发现在碱性N-甲基哌啶存在下,由简单的铁盐和硫生成的Fe/S簇非常有效地催化硝基芳烃与芳基甲基氯化物之间的氧化还原缩合反应。反应以中等至良好的产率进行,具有良好的功能耐受性。
由于苯并咪唑和苯并噻唑在有机电子材料,生物活性天然产物特别是药物化合物中的广泛存在,它们是有价值的结构单元[1,2] 制备它们的常用方法涉及1,2-二氨基苯或2-氨基硫酚与在高温或氧化条件下醛、醇、羧酸或苄胺的缩合反应[3,4] 邻苯二胺被用作这些反应的底物。然而,在这些转化中尚未报道由邻硝基芳烃和芳基甲基氯直接一锅合成苯并咪唑或苯并噻唑。
近年来,硝基与其他还原组分的氧化还原缩合为含氮化合物提供了直接、分步、原子和氧化还原经济的方法,引起了广泛关注[5] 例如,Wang和合作者[6] 已经报道了金纳米颗粒催化邻硝基苯酚/苯胺与醇之间的氧化还原缩合 [方案1,等式。(1)]。Nguyen等人[7]开发了Fe/S催化的邻硝基苯胺与苄醇、苄胺或芳酸的环化反应 [(方案1,等式(2)],诸如此类反应[8] 即便如此,探索一种由简单易得的起始原料合成2-取代的苯并恶唑的新方案仍然迫切。作为延续我们对硫介导的反应的兴趣,[9] 在这里我们报道Fe/S催化邻硝基芳烃与芳基甲基氯化物的环化反应制备苯唑的简单有效方法(方案1)。
选择N-甲基-2-硝基苯胺(1a)和苄基氯(2a)作为模型底 物,其中FeCl3·6H2O(5 mol%)和S8(40 mol%)探索反应。筛选了不同的参数。有机碱,例如三乙胺(TEA),N-甲基吗啉、吡啶和2-甲基吡啶产生痕量产物(表1,条目1-4)。然后我们以N-甲基哌啶为碱和溶剂探索反应。令人惊讶的是,在130 8C下20 h以80%的产率获得了产品3a(表1,条目5)。此外,当使用一当量的S8时,我们以90%的产率获得了3a(表1, 条目6)。将反应温度提高到150 oC不会提高产物的收率(表1, 条目7),而将温度降低到110 oC,则会显着降低收率(表1, 条目8)。用FeCl2·4H2O代替FeCl3·6H2O的收率较低(表1,第9项)。此外,改变BnCl和硫的当量数并不能改善反应(表1,条目10-12)。当用BnBr代替BnCl时,获得了类似的结果(表1, 条目13)。另外,在空气中进行反应导致较低的收率(表1, 条目14)。最佳反应条件为:N-甲基-2-硝基苯胺(1.0当量),BnCl(1.2当量),FeCl3·6H2O(5摩尔%),S8(1.0当量)于130 oC在N-甲基哌啶(2 mL)中反应20小时。
方案1. 用于合成苯并唑的最新氧化还原策略。
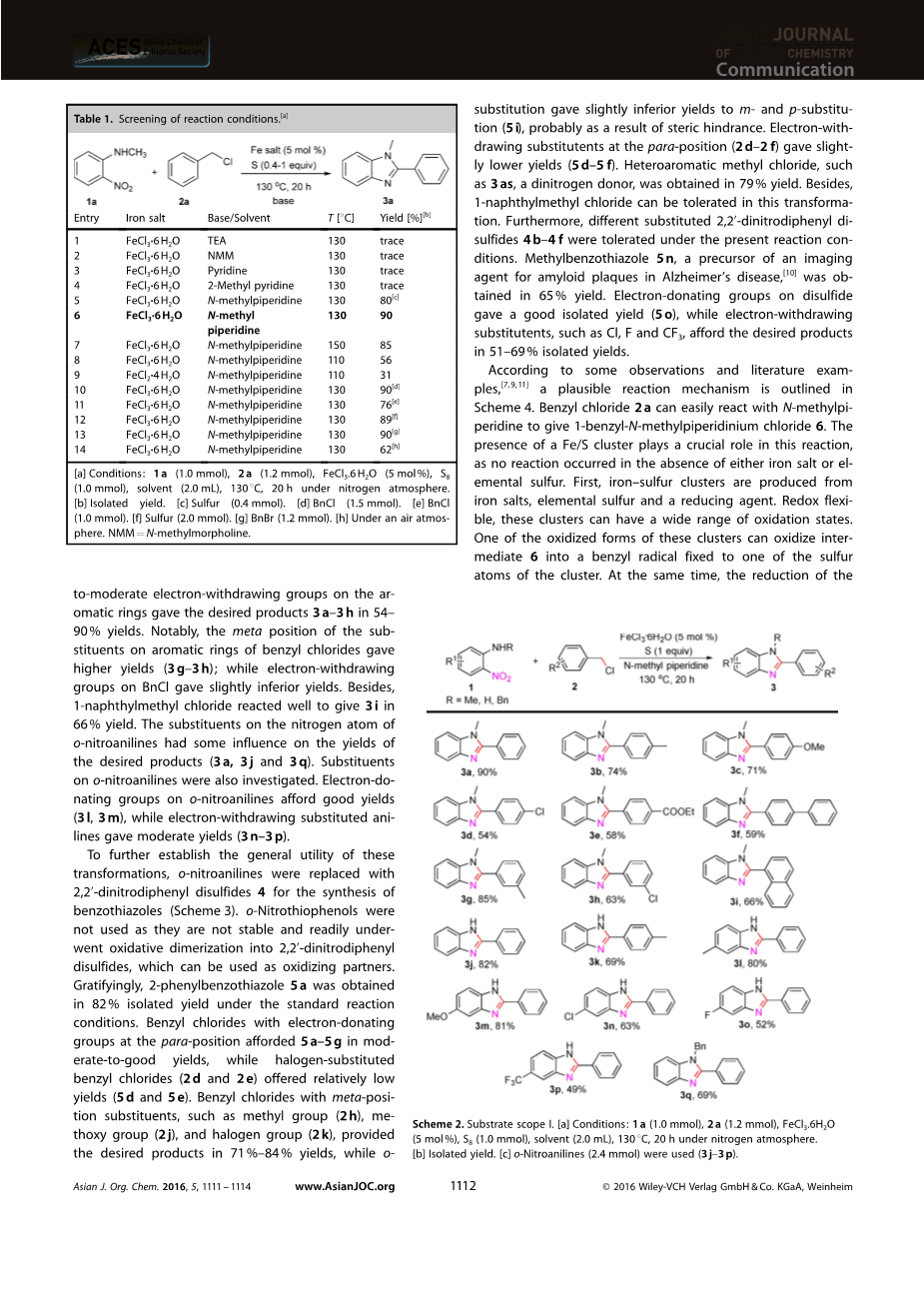
方案2概述了上述优化条件对各种取代苄基氯和邻硝基苯胺衍生物的适用性。芳香环上供电子或低至中度吸电子基团的芳甲基氯以54-90%的收率得到所需的产物3 a-3h。值得注意的是,苄基氯的芳环上取代基的间位具有更高的产率(3 g–3 h); 而BnCl上的吸电子基团的收率稍差。此外, 1-萘甲基氯反应良好,以66%的收率得到3i。邻硝基苯胺的氮原子上的取代基对所需产物(3a,3j和3q)的产率有一定影响。还研究了邻硝基苯胺上的取代基。邻硝基苯胺上的给电子基团提供良好的收率(3 l,3 m),而吸电子的取代苯胺给出中等的收率(3 n–3 p)。
为了进一步确立这些转化的通用性,用2,2-二硝基二苯基二硫化物4取代了邻硝基苯胺,用于合成苯并噻唑(方案3)。不使用邻硝基硫代苯酚, 因为它们不稳定并且容易进行氧化二聚为2,2-二硝基二苯基二硫化物,可以用作氧化伙伴。令人欣慰的是,在标准反应条件下,以82%的分离产率获得了2-苯基苯并噻唑5a。在对位带有给电子基团的苄基氯以中等至良好的产率提供5 a-5g,而卤素取代的苄基氯(2d和2e)提供相对较低的产率(5d和5e)。具有间位取代基的苄基氯,例如甲基(2 h),甲氧基(2 j)和卤素基团(2 k),以71%–84%的收率提供了所需的产品,而邻位取代可能比间位和对位取代的收率稍低(5 i),这可能是由于空间位阻的影响。在对位(2 d–2 f)处的吸电子取代基产生的产率稍低(5 d–5 f)。诸如3as这样的杂芳族氯甲烷(二氮供体)以79%的产率获得了所需产物。此外,1-萘甲基氯在该转化中可以耐受。另外,在目前的反应条件下,不同的取代的2,2-二硝基二苯基二硫化物4 b–4f可以耐受。甲基苯并噻唑5 n-阿尔茨海默氏病中淀粉样蛋白斑的显像剂的前体,可以65%的收率获得[10]。二硫化物上的给电子基团提供了良好的分离产率(5 o),而吸电子取代基(如Cl、F和CF3)以51-69%的分离产率提供了所需的产物。
根据实验观察和已报道的文献[7,9,11],方案4中概述了一个合理的反应机理。苄基氯2a可以轻松地与N-甲基哌啶反应生成1-苄基-N-甲基哌啶鎓氯化物6。Fe/S簇在该反应中起关键作用,因为在不存在铁盐或元素硫的情况下没有反应发生。首先,铁硫簇是由铁盐、元素硫和还原剂产生的。这些簇由于具有氧化还原柔性,可以具有广泛的氧化态。这些簇的氧化形式之一可以将中间体6氧化成连接在簇的硫原子之一上的苄基自由基。同时,Fe/S团簇还原形式之一可还原N-甲基邻硝基苯胺1a 的硝基。由于氧化还原的柔韧性和酸碱性质,Fe/S簇催化了随后的氧化还原和缩合步骤,形成了最终产物2-苯基-N-甲基苯并咪唑3a。
总之,我们已经开发出由Fe/S催化的邻硝基芳烃与芳基甲基氯化物的环化反应生成苯并唑的氧化还原缩合方法。反应以中等至良好的产率进行,具有良好的功能基耐受性。初步的机理研究表明,铁硫簇参与了反应。该反应简单、直接、步骤和氧化还原经济,这使该方法具有吸引力和实用性。未来的研究将集中于研究范围、详细的反应机理以及对生物学上感兴趣的分子的扩展。
方案2. 底物范围I. [a]条件:1a(1.0 mmol),2a(1.2 mmol),FeCl3.6H2O(5 mol%),S8(1.0 mmol),溶剂(2.0 mL),130 oC,氮气氛下20小时。[a]分离的收率。[c]使用邻硝基苯胺(2.4 mmol)(3j–3p)。
方案3. 底物范围II。[a]条件:4a(0.5 mmol),2a(1.2 mmol),FeCl3.6 H2O(5 mol%),S8(1.0 mmol),溶剂(2.0 mL),130在氮气氛下于130 oC,20 小时。[b]分离的收率。[c] 2-氯甲基吡啶盐酸盐用作起始原料。
方案4.合理的反应机理。
实验部分
合成苯唑的一般步骤:邻位硝基亚芳基1(1.0 mmol或2.4 mmol) 或4(0.5 mmol)的混合物,将芳基甲基氯 2(1.2 mmol), 元素硫(1.0 mmol),FeCl3.6H2O(5 mol%,0.05 mmol)和N-甲基哌啶(2 mL)放入装有磁力搅拌棒的密封压力容器(25 mL)中然后加盖并在氮气氛下于1308℃搅拌20小时。反应完成(TLC)后,将混合物冷却至室温,用甲醇(2 mL)和DCM(15 mL)稀释,真空蒸发, 并通过硅胶柱色谱法纯化,使用石油醚和乙酸乙酯作为洗脱剂得到所需的苯并唑类化合物。
致谢
感谢中国国家高技术研究发展计划(No. 2013AA031901)的资助。
关键字:芳基甲基氯化物 苯并咪唑
苯并噻唑 元素硫 铁盐
[1] a) Z. Zhu, B. Lippa, J. C. Drach, L. B. Townsend, J. Med. Chem. 2000, 43, 2430 – 2437; b) P. Chaudhuri, B. Ganguly, S. Bhattacharya, J. Org. Chem. 2007, 72, 1912 – 1923; c) M. L. Morningstar, T. Roth, D. W. Farnsworth, M. K. Smith, K. Watson, R. W. Buckheit, Jr., K. Das, W. Zhang, E. Arnold, J. G. Julias, S. H. Hughes, C. J. Michejda, J. Med. Chem. 2007, 50, 4003 – 4015; d) L. Hu, M. L. Kully, D. W. Boykin, N. Abood, Bioorg. Med. Chem. Lett. 2009, 19, 3374.
[2] a) W. S. Saari, J. M. Hoffman, J. S. Wai, T. E. Fisher, C. S. Rooney, A. M. Smith, C. M. Thomas, M. E. Goldman, J. A. Orsquo;Brien, J. Med. Chem. 1991, 34, 2922 – 2925; b) A. D. Westwell, A. A. Weekes, Curr. Med. Chem. 2009, 16, 2430 – 2440; c) M. M. M. Raposo, M. C. R. Castro, M. Belsley, A. M. C. Fonseca, Dyes Pigm. 2011, 91, 454 – 465; e) A. Banerjee, S. K. Santra, S. Guin, S. K. Rout, B. K. Patel, Eur. J. Org. Chem. 2013, 1367 – 1376; f) M. Feng, B. Tang, S. H. Liang, X. Jiang, Curr. Top. Med. Chem. 2016, 16, 1200 – 1216.
[3] a) H. Sharma, N. Singh, D. O. Jang, Green Chem. 2014, 16, 4922 – 4930; b) A. J. Blacker, M. M. Farah, M. I. Hall, S. P. Marsden, O. Saidi, Org. Lett. 2009, 11, 2039 – 2042; c) O. D. Thomas, T. J. Peckham, S. Holdcroft, J. Am. Chem. Soc. 2012, 134, 10753 – 10756; d) G. Naresh, R. Kant, T. Narender, J. Org. Chem. 2014, 79, 3821 – 3879.
[4] a) T. B. Nguyen, L. Ermolenko, W. A. Dean, A. Al-Mourabit, Org. Lett. 2012, 14, 5948 – 5951; b) T. Guntreddi, R. Vanjari, K. N. Singh, Org. Lett. 2014, 16, 3624 – 36
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[255336],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 选择性能量转移催化烯烃的含硼几何异构化外文翻译资料
- 瑞德西韦阻滞SARS-CoV-2聚合酶的作用机理外文翻译资料
- 铱催化的共轭二烯的C-H烯基的烯丙基化反应外文翻译资料
- 铱和布朗斯特酸协同催化烯丙醇对萘酚衍生物的对映选择性脱芳构化外文翻译资料
- 新型选择性MT2受体配体2-(苯硫基)苯并[b]噻吩类化合物的制备和药理学评价外文翻译资料
- Ilimaquinone是一种海绵代谢产物,通过gadd153介导的途径发挥抗癌作用外文翻译资料
- 用环境敏感药物释放的三氧化二砷靶向介孔二氧 化硅纳米颗粒有效治疗三阴性乳腺癌外文翻译资料
- 复方中草药对雄性荷斯坦犊牛生长表现,胴体特征和肉质的 影响外文翻译资料
- 线粒体在没有人体ATP合酶的亚基c时渗透性转 变的持久性外文翻译资料
- 基于呋喃类化合物构建环氧树脂 2,5-呋喃羧酸(FDCA)生物基环氧树脂的合成及性能研究外文翻译资料

