碳化硼纳米线修饰双功能阴极衬底的长寿命锂硫电池
作者
Liu Luo, Sheng-Heng Chung, Hooman Yaghoobnejad Asl, and Arumugam Manthiram*
摘要
开发高能量密度锂硫(Li-S)电池依赖于电极基板的设计,这种基板既能承载高硫负荷,又能实现高电化学利用率。本文介绍了一种新型的双功能阴极衬底,该衬底通过一个简单的催化辅助过程在碳上原位生长了硼碳化物纳米线构成纳米纤维(B4C@CNF)。作为化学锚定中心的B4C纳米线具有较强的多硫化物吸附能力,实验数据和第一性原理计算验证了这一点。同时,B4C的催化作用也加快了多硫化物转化的氧化还原动力学,有助于提高反应倍率性能。以B4C@CNF为阴极底物的电池,经过500次循环后,可显著保持80%的效率,并在4C速率下实现稳定的循环能力。此外,B4C@CNF衬底使阴极能够同时实现高硫含量(70% wt%)和高硫负荷(10.3 mg cmminus;2),提供了9 mAh cmminus;2的卓越面积容量。此外,使用B4C@CNF衬底制备的Li-S囊炮,每个阴极可容纳200 mg的高硫质量,经过50次循环后可提供125 mAh的高放电容量。
正文
电动汽车和便携式电子设备市场的蓬勃发展,推动了储能行业探索比传统锂离子电池技术具有更高能量密度和更低成本的先进充电电池。在众多竞争者中,锂硫电池因其高理论容量(1675毫赫gminus;1)、丰富的自然资源和低成本的环保硫引起了人们的特别关注。然而,阻碍Li-S技术商业化的挑战依然艰巨。例如,硫及其排放产物本身的导电性较低,导致活性物质利用有限,反应动力学缓慢。同时,作为氧化还原中间体形成的可溶性聚硫化物容易溶解在液体电解质中,从阴极向阳极区域不可逆扩散,导致电化学不稳定,循环寿命较差[1,4]。
鉴于这些缺点,人们对电池结构的合理设计和工程设计进行了大量的工作。[5,6]特别是开发优质硫阴极对于实现高能量密度、长周期寿命的承诺具有重要意义。[3,7]阴极设计中最常用的方法之一是利用导电碳质材料作为物理屏障来捕获聚硫化物。然而,非极性碳质寄主的物理约束可能不足以保证极性聚硫化物在较长寿命内受到抑制扩散,特别是对于高负荷电池的设计。[4,7]针对这一问题,提出了一种利用功能极性基板作为高效硫载体的锂电池替代策略。例如,过渡金属氧化物、硫化物、碳化物和氮化物作为硫底物的应用,通过引入路易斯酸碱相互作用,[8,9]形成强化学键,[10-12]或对扩散聚硫化物起到催化作用,极大地提高了聚硫化物的保留能力。[13-15]近年来,过渡金属碳化物如TiC、[16,17]Ti2C、[8]Mo2C、[18]W2C、[18]、WC[17]等已被应用于硫阴极构型,并被证明对改善锂电池的电化学功能是有效的。然而,它们中的大多数都有一个严重的缺点,低硫含量(lt; 56% wt%)和低硫负荷(lt; 2mg cmminus;2)。[8,17]通常采用叶片铸造法,将碳化物与额外的炭黑和粘结剂(如聚偏氟乙烯)混合制成硫基板,然后浇铸到集电器上。这些惰性添加剂和集电体在阴极中占相当大的比重。此外,大多数碳化物的密度比硫高得多,这也限制了硫含量的可达性。[17,18]因此,有必要探索具有粘结剂/电流无集电极制备策略的轻质高效材料,以提高聚酰亚胺的保留能力,实现可接受电化学性能的Li-S电池。
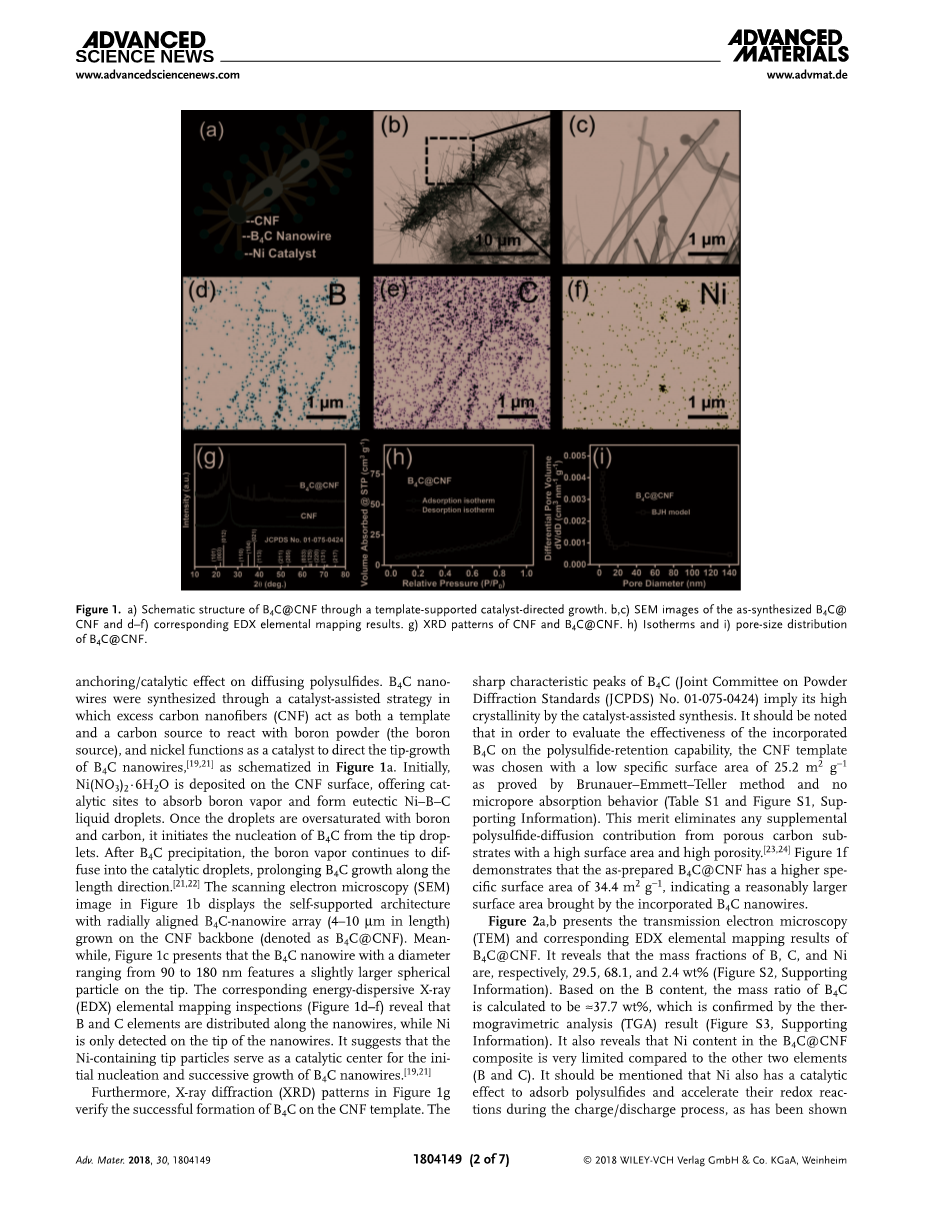
碳化硼(B4C)由于其低密度(asymp;2.5 g cmminus;3),[19]良好的导电性(1.25-3.33 S cmminus;1),[20]以及优异的催化效果,是一种很有前途的电池应用候选材料[20,21]。本文首次将轻质B4C基板成功地应用于Li-S系统中,通过简单、可伸缩的制造工艺,证明了该基板对聚酰亚胺的扩散具有良好的锚定/催化作用。B4C catalyst-assisted策略的方法制备了纳米线中过量的碳纳米纤维(CNF)作为模板和碳源与硼粉反应(硼源)和镍作为催化剂直接B4C纳米线的生长,[19、21]如图1个图示。一开始,在CNF表面沉积Ni(NO3)2∙6H2O,提供吸收硼蒸气并形成共晶Ni - b - c液滴的催化位点。当液滴被硼和碳过饱和后,就会从液滴的尖端开始形成B4C的成核。B4C析出后,硼蒸汽继续扩散到催化雾滴中,B4C沿长度方向生长时间延长。[21、22]扫描电子显微镜(SEM)图像如图1 b显示且架构与径向对齐B4C-nanowire数组(4到10mu;m长)生长在CNF骨干(表示B4C@CNF)。同时,从图1c中可以看出,直径为90 - 180 nm的B4C纳米线,其尖端的球形颗粒略大。对应的能量色散x射线(EDX)元素映射检查(图1d-f)显示,B和C元素沿纳米线分布,而Ni只在纳米线的尖端检测到。结果表明,含ni的针尖粒子是B4C纳米线初始成核和后续生长的催化中心。[19,21]此外,图1g中的x射线衍射(XRD)图谱验证了B4C在CNF模板上的成功形成。B4C (JCPDS) 01-075-0424号粉末衍射标准联合委员会(JCPDS)的显著特征峰表明,通过催化辅助合成,B4C具有较高的结晶度。应该注意的是,为了评估的有效性结合B4C polysulfide-retention能力,CNF模板选择的低比表面积25.2平方米克minus;1 Brunauer-Emmett-Teller证明方法和没有微孔吸附行为(表S1和图S1,支持信息)。这一优点消除了来自高表面积和高孔隙率多孔碳基板的任何补充聚砜扩散贡献。[23,24]图1f显示,制备的B4C@ cnf比表面积更高,为34.4 m2 gminus;1,说明B4C纳米线的加入带来了更大的比表面积。
图2a,b为透射电镜(TEM)和对应的B4C@CNF的EDX元素映射结果。结果表明,B、C、Ni的质量分数分别为29.5、68.1、2.4 wt%(图S2,支持信息)。根据B含量计算出B4C的质量比约为asymp;37.7 wt%,热失重分析(TGA)结果证实了这一点(图S3,支持信息)。它还表明,镍含量B4C@CNF复合其他两个元素相比非常有限(B和C)。它应该提到倪也有催化作用吸附聚硫化物和加速氧化还原反应在充电/放电过程中,如同shownin我们之前的工作。然而,由于B4C@ cnf中Ni的含量有限,与B4C相比,Ni的催化作用微不足道。另一方面,在图2c中,B4C纳米线所选择的区域电子衍射(SAED)图形被索引到一个三角晶体系统中,晶格参数与B4C的参数完全匹配(a = 5.6 a, c = 12.12 a)。此外,同一晶体的高分辨率透射电镜(HRTEM)图像显示了相应的晶格条纹到d101平面间距(图2d)。将SAED得到的互反晶格矢量叠加在实空间TEM显微图的顶部,我们可以确定[- 103]为B4C纳米线的生长方向。据此,重建了B4C的一个初步纳米线模型,该模型包含沿生长方向侧向暴露的(100)面和(110)面以及顶部和底部的三倍(103)面(图2e)。
合成的B4C@CNF复合材料分散在异丙醇溶液中,真空过滤成薄膜作为独立的阴极衬底。这种集成的B4C@CNF基板具有多种优点,可以改善Li-S电池的性能。首先,亲硫B4C纳米线为化学吸附聚硫化物提供了锚定位点,从而有效地将聚硫化物限制在B4C和CNF网络中。只有当局域聚硫化物吸附在导电衬底表面时,才能触发其氧化还原反应。其次,B4C的催化作用可以降低过电位,促进多硫化物转化的氧化还原动力学。具体来说,在放电过程中,B4C可以加速长链Li2Sx (4 lt; xle;8)还原为最终产物短链Li2S/Li2S2,并沉积在B4C纳米线周围区域。同样,在充电过程中,这些沉积的短链Li2S/Li2S2会吸附在B4C表面,由于B4C的催化能力,这些沉积的短链Li2S/Li2S2会迅速氧化为长链Li2Sx (4 lt; xle;8)。由于化学平衡的变化,这种促进的氧化速率会进一步加速2s的初始活化。B4C@CNF基板除了具有更好的聚硫吸附和聚硫转化能力外,作为一种独立的结构,还不使用额外的集电体和惰性粘结剂。它极大地减少了非活性物质的质量和潜在的电极降解,[25]保证了70% wt%的高硫含量,同时仍然保证了优良的电池性能。
为了探究B4C对聚硫醚的吸附能力,我们进行了聚硫醚吸附实验,如图3a所示。以B4C@CNF和CNF为吸附剂,分别浸在含0.5times;10minus;3 m Li2S4的多硫化物溶液中,置于1,3-二甲苯/二甲氧基乙烷中。有趣的是,在短暂的静置30分钟后,B4C@CNF对Li2S4溶液进行了彻底的脱色,而CNF样品中仍然保留了Li2S4的黄色。这种直观直观的对比表明,B4C具有明显的聚硫锚固效果,优于纯CNF对聚硫化物的弱物理吸附作用。此外,为了更深入地了解B4C对化学多硫化物吸附的显著性,采用密度泛函理论进行理论计算,模拟了B4C的结合几何和能量。根据前面的讨论,B4C纳米线暴露了不同的面(图2e),选择B4C的不同代表性面(100)、(110)、(001)和(111)与Li2S4相互作用,获得了更全面的Li2S4 - B4C相互作用的概况。并对石墨基面进行了研究。结果表明,只有非极性C-C键的石墨对Li2S4的结合能为1.18 eV(图S4,支撑信息)。对比之下,Li2S4和B4C的结合能要高得多,根据表面劈理的性质和方向,其结合能在3.84 ~ 12.51 eV之间(图3b)。实验结果表明,B4C明显提高了多硫化物的吸附性能,支持了上述吸附实验结果。此外,对不同B4C方面的结合能变化进行更深入的考察,揭示了有关具有吸引力的Li2S4-B4C相互作用性质的更深刻的事实。如图3d所示,当接近(110)面和(001)面时,Li2S4仍然保持S-S连接,分别贡献了3.84 eV和5.84 eV的结合能。然而,当涉及到(111)和(100)面时,S-S键断裂,随后在B4C衬底的S和表面原子之间形成新的键。此外,对成键模式的分析表明,Li2S4-B4C相互作用的大小与形成的S-B键的数量呈正相关。例如,有4个S-B键的(100)面具有最高的结合能12.51 eV,而没有S-B键和2个S-C键的(110)面具有最低的结合能3.84 eV。此外,很明显,S-C键的形成不如S-B键有利,因为添加S-C键会相对降低结合能(表S2,支撑信息)。
为了更详细地了解Li2S4和B4C之间的相互作用,我们进行了巴德电荷分析,定量揭示了这些原子之间的电子电荷转移。这里的电荷差是指被吸附和被分离的Li2S4-B4C体系之间原子电子密度的变化。如图3c所示,结合能的大小与S原子的巴德尔电荷关系很好。最初,在一个孤立的Li2S4分子中,每个S原子(记作B (S))的平均巴德尔电荷为- 0.45,接近其经典氧化态(- 0.50)。当吸附在(111)和(100)面上时,Li2S4的B (S)值的负值更多(分别为- 0.66和- 1.29 eV),这表明部分还原的S原子以最初分离的Li2S4形式进一步还原。对于(001)面和(110)面,B (S)的值变得更加正值(分别为- 0.11和- 0.036 eV),这意味着S原子被表面C原子部分氧化,相对于前者的结合能相对较低。[26,27]基于S、B和C原子的相对电负性,以及不协调表面C原子的氧化作用,这些观察结果可能是合理的。此外,所有B和C原子组成的面(表S3,支持信息)的Bader电荷的总和可以让我们了解当Li2S4吸附在B4C衬底上时,电子流的方向。值得注意的是,通过绘制被吸附的Li2S4-B4C体系与原子密度叠加之间的电荷密度差,也可以观察到基本的电子电荷转移(图3e)。其中蓝色和红色的表面轮廓分别代表吸附后电荷的获得和丢失的空间区域。这为吸附后S与B4C表面原子之间的电子密度增加提供了直接证据(蓝色部分),说明了Li2S4与B4C之间形成了化学键。[26,27]理论分析验证了B4C具有显著的聚硫吸附能力,其结合能可达12.51 eV。事实上,这一数值甚至高于其他报道的涉及路易斯酸活性底物的研究,如过渡金属硫系化合物(TMCs) (3.5-7.0 eV)。[9],它可能源于B4C的氧化还原性质更活跃和差旅管理公司相比,鉴于在差旅管理公司往往低氧化态的过渡金属原子不可能改变了聚硫的氧化还原性质及其保留主要依赖于纯粹的酸碱相互作用。[28、29]
图4a显示了以CNF和B4C@ CNF为阴极衬底,不同硫阴极的纽扣电池循环伏安(CV)剖面。所记录的CV曲线由两个阴极峰(1.9-2.3 V)和一个宽阳极峰(2.4-2.5 V)组成,分别表示硫还原为多硫化物后再还原为硫化锂混合物(Li2S2/Li2S)[30,31]和反氧化反应(Li2S2/Li2S还原为Li2S8/S)。[32,33]使用B4C@CNF的电池与使用原始CNF的控制电池相比,峰值电流更高,阴极和阳极峰分离更小,表明B4C的催化作用加快了多硫化物转化的氧化还原动力学。[15,18,34]此外,在含有两个相同电极和Li2S6溶液作为电解质的对称电池中,CV试验也验证了B4C的催化能力。作为参考,我们还进行了不含li2s6电解质的对称电池,以消除CV中电容电流的影响,结果发现,电容电流对电流的贡献接近于零。[14]15 mV sminus;1的扫描速率(图S5,支持信息),它揭示了简历,当前单元中采用B4C@CNF电极远高于控制电池的使用CNF,指示的增强聚硫化物的氧化还原动力学转换带来的B4C的催化效果。当扫描速率降低到3mv sminus;1时,与对照组相比,使用B4C@CNF的电池中可以观察到电流更高的氧化还原峰,这表明电化学可逆性较好,并促进了多硫化物的转化。此外,锂离子扩散系数(DLi )值在阳极(A1)和阴极(C1, C2)的山峰与B4C@CNF电池,分别1.3times;10minus;7,2.4times;10minus;8和1.6times;10minus;7厘米2 sminus;1(图S6,支持信息),也高于传统的参考价值Li-S电池(2times;times;10 - 10minus;minus;9厘米2 sminus;1)。这种促进锂离子扩散的作用,再次证实了B4C在充放电过程中的催化作用,使电化学环境更加有利,氧化还原动力学加速。
此外,B4C@CNF中氧化还原化学环境的改善也体
全文共11969字,剩余内容已隐藏,支付完成后下载完整资料

英语原文共 7 页,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[1840]
您可能感兴趣的文章
- BaTiO3和SrTiO3纳米立方单晶体的有 序组装的压阻响应特性外文翻译资料
- 结构对有机硅改性酚醛树脂热稳定性及抗氧化机理的影响外文翻译资料
- 磷酸三(2-巯基乙基)固化环氧热固树脂的高折射率和阻燃性外文翻译资料
- 燃烧合成TiB2-Cu金属陶瓷的抗烧蚀性外文翻译资料
- 氢键在光诱导水离解中的作用:一把双刃剑外文翻译资料
- 碳酸氢钠/偶氮二异丁腈协同作用对低密度不饱和聚酯树脂制备的影响外文翻译资料
- 利用钢渣和草酸氢钾制备新型化学键合陶瓷外文翻译资料
- A位空位型钛酸铋钠基弛豫铁电体 具有超高的能量密度和更高的放电效率外文翻译资料
- 用热分析方法测定含氯化钠和氯化钾的油 井水泥浆体的水化产物外文翻译资料
- 将垂直排列的石墨烯片多孔膜用于高效太阳能热净水外文翻译资料

