
英语原文共 8 页,剩余内容已隐藏,支付完成后下载完整资料
新型多点相转移催化剂的合成与表征茚分子内环戊基化动力学研究
从低成本的原料中可以很容易地获得迄今未知的新型多点催化剂,即1,3,5-三(苄基三乙基溴化铵)苯(TBTABB)。通过1H NMR, 13 CNMR,IR和元素分析证明合成的MPTC和螺茚衍生物的结构。通过使用过量的氢氧化钠水溶液和1,4-二溴丁烷,在假一级条件下遵循茚的环戊烷化动力学证明了新的多位点相转移催化剂的潜力。茚的环戊烷化动力学在低温(40℃)下进行,并通过气相色谱监测。此外,通过研究各种环戊烷化反应的动力学,将TBTABB的催化效率与普通单体中心催化剂的催化效率进行比较。在研究六种相转移催化剂时,我们发现TBTABB是高效的。研究考虑了各种实验参数和条件的影响,包括搅拌速度,催化剂浓度,底物浓度,氢氧化钠浓度和温度对反应速率的影响,研究了多点相转移催化剂对反应速率的影响。来自Arrhenius图,发现活化14.18kj/mol 。还评估了热力学参数如熵变、焓变和活化能。利用动力学和热力学结果,我们提出了茚的环戊烷化的界面机制。从不同的实验可以得出结论,TBTABB作为多点相转移催化剂具有很大的潜力。
1. 介绍
绿色化学在有机合成中日益增长的重要性推动了环境友好的新合成方法的发展.鉴于近年来日益增加的环境和经济问题,现在化学家必须尽可能多地发现环境友好的催化反应。为了成功完成涉及亲脂反应物和亲水反应物的反应,必须尽可能经常地使反应物彼此碰撞。但是必须注意到反应物本质上是不混溶的。如果在这种系统的严苛条件下使用质子溶剂,可以提高转化率,但通常伴随着一些不利的副反应.结果,这些技术在工业上没有吸引力,受到约束和污染。
现在广泛称为“相转移催化”(PTC)的合理技术被开发用于克服遭遇由于水相与有机相的相互不溶性导致的难题.在这个关键的绿色方法存在于不混溶相的化学反应物,相转移催化剂,具有通过穿透界面将一种反应物作为高活性物质携带到发生反应的另一相中的能力。使用相转移催化的主要优点是在中等反应条件下获得大转化率,高反应速率和良好选择性。如今,PTC成为有机合成的重要选择,并广泛应用于特种化学品的制造过程,如药品,染料,香水,润滑剂添加剂,农药和聚合物合成用单体。PTC方法是用于构建碳 - 碳键的最通用和常用的反应之一。Jarrouse首次报道了活性腈与PTC下的氢氧化钠水溶液的烷基化反应并由Makosza和Jonczyk进行了非常详细的调查[18].在各种PTC中,铵盐因其可用性和易于反应后处理而备受关注。然而,目前,在相转移催化剂的选择中,鎓盐的规模经济和效率具有重要意义,特别是对于明确的工业规模制备。有机化合物。随着使用的增长,人们对开发具有更高催化效率的相转移催化剂付出了很多努力。为此,开发了“多点”相转移催化剂(MPTC)。与单中心盐相比,这种鎓盐的显着特征包括容易制备,低能量需求和在温和反应条件下的特定合成转化中的高反应性。
Idoux等首次合成了可溶和不溶形式的多点相转移催化剂(MPTC),它在含有两个可取代基团的有机试剂的置换中表现出更高的选择性。Benaglia等[20] 和Murugan和Gopinath[21] 已经报道了使用聚合物支持的多点相转移催化剂对吡咯进行N-烷基化。王和谢[22] 合成了1,4-双(三乙基甲基铵)二氯化苯,并通过遵循4-乙烯基-1-环己烯的二氯环丙烷化反应动力学来研究其效率。最近,阿里[23] 报道了一种通过N-烷基化合成叔胺的有效方法a,v-二卤代烷烃使用多位点相转移的胺衍生自氢醌的催化剂。早些时候,我们报道了加入二氯卡宾的不同MPTC[24,25] 和Darzen的凝结[26].我们最近比较了[27] 具有单中心相转移催化剂的MPTC在苯基乙腈烷基化中的效率。由于相当可观随着催化活性的增加,MPTC在化学家中变得越来越有吸引力。
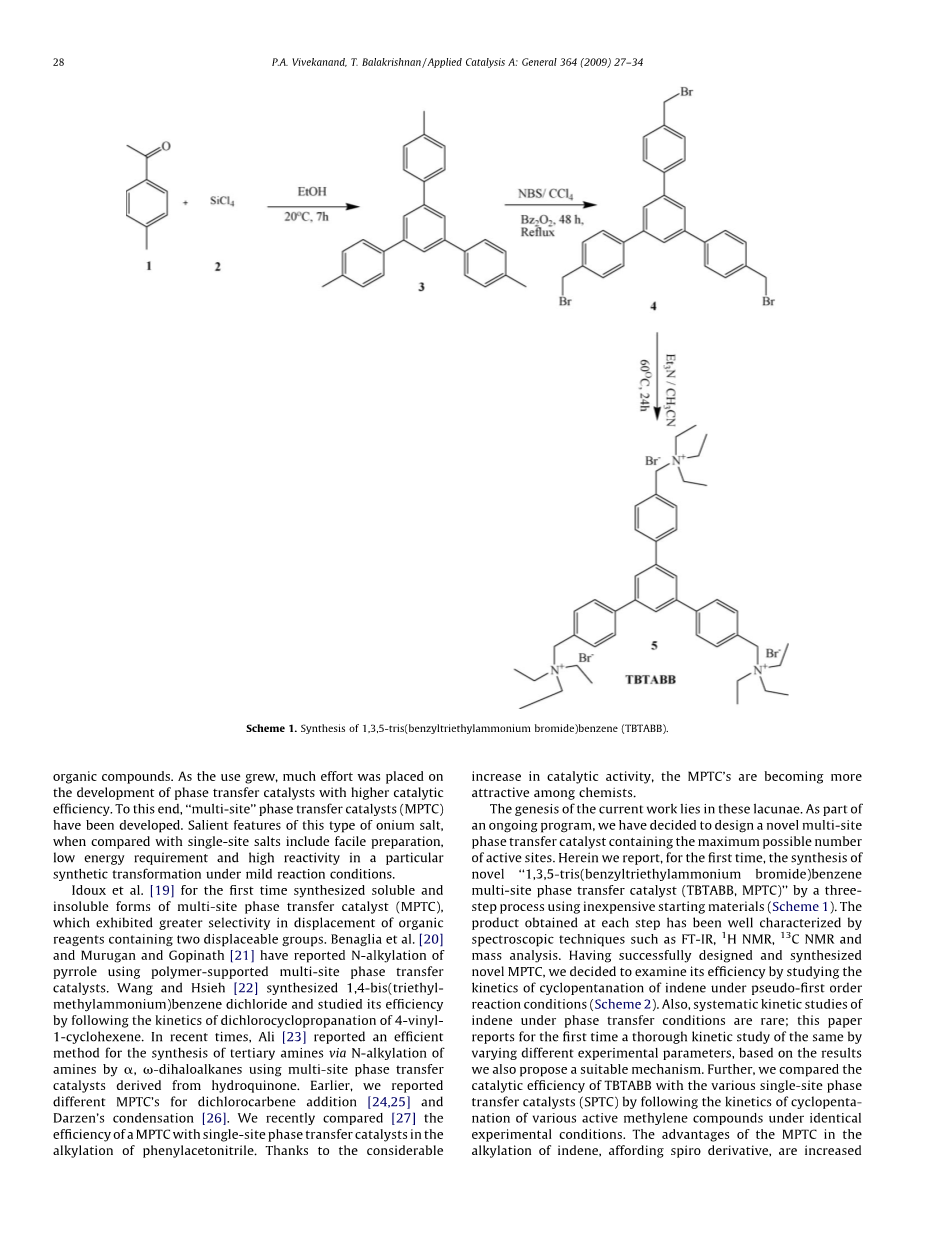
当前工作的起源在于这些空白。作为正在进行的计划的一部分,我们决定设计一种新型多点相转移催化剂,其中含有尽可能多的活性位点。在这里,我们首次报道了使用廉价原料通过三步法合成新型1,3,5-三(苄基三乙基溴化铵)苯多点相转移催化剂(TBTABB,MPTC)(方案1).通过光谱技术如FT-IR,1 H NMR, 13 C NMR和质谱分析充分表征了在每个步骤获得的产物。在成功设计和合成新型MPTC后,我们决定通过研究在伪一级反应条件下茚的环戊烷化动力学来研究其效率(方案2).此反应速率和选择性,亲水条件和低能量要求。
2. 试验
2.1. 化学制品
对甲基苯乙酮( Merck ),1,4- 二溴丁烷( Lancaster ) ,茚(Acros),苯乙腈(Lancaster),氢氧化钠(SRL),氯仿(SRL),三乙胺(SRL),丁二酰亚胺 (SRL),过氧化苯甲酰(SRL )和十六烷(Merck)是从商业来源采购的并且原样使用
2.2. 仪表
在Bruker-Tensor 27 FT-IR分光光度计上记录IR光谱。分别使用JEOL 400MHz和JEOL 500MHz光谱仪记录1 1 H NMR和 13 13 C NMR光谱。在Jeol-JMS-DX 300 HF质谱仪上记录质谱。在PerkinElmer 240B元素分析仪上进行元素分析。在具有火焰离子化检测器和Vista CDS 401数据站的Varian 3700型上进行气相色谱分析。使用的柱是5%SE-30 ChromWHP 80 / 100,3M 1/8英寸不锈钢管。
2.2.1. 1,3,5-三(4-甲苯基)苯的合成
向搅拌的对甲基苯乙酮溶液中加入1(1.5克,在20℃下,使用注射器缓慢加入11.17mmol)的无水乙醇(20ml)溶液,SiCl 4 (1.89g,11.17mmol)。7小时后,将反应混合物倒入水(40ml)中,用CH 2 Cl 2 (2times;25ml)萃取,干燥(MgSO 4 4 ),过滤并真空浓缩。最后,将由此获得的粗产物从乙醇中重结晶。熔点176-177℃,收率80%,IR(KBr),cm-1 :1470,1674,2962;1 1 H NMR(400MHz,CDCl3 ):d 2.45(s,9H),7.29(d,6H,j = 7.8 hz)、7.62 (d, 6h, j = 8.2 hz)、7.62 (s, 3h);13 c 核磁共振 (100 兆赫,CDCl 3 ):d 25.3,129.8,129.6,133.5,137.3,125.2,137.5。
2.2.2. 1,3,5-三[4-溴甲基苯基]苯的合成
将新鲜重结晶的N-溴代琥珀酰亚胺(4.29g,24.0mmol)分7次加入,间隔7小时,得到1,3,5-三 - (4-甲基苯基)苯(2.6g,7.5mmol)在CCl中的溶液(回流后加入 4 (400ml),每次加入后立即加入过氧化苯甲酰(80mg)。在回流的总反应时间49小时后,冷却各混合物并通过过滤除去沉淀的琥珀酰亚胺。然后在真空中完全除去溶剂,得到粗制溴化合物,为固体。使用己烷:氯仿(4:1)作为洗脱液对粗产物进行色谱分离(SiO 2 ),得到纯的三溴衍生物。熔点193-194℃,收率81%,FT-IR:KBr,cm-1 :775, 1413, 1688, 2960;1 1 H NMR(500MHz,CDCl3 ):d 4.5(s,6H,-CH 2 Br),7.49(d,J = 6.9Hz,6H),7.61-7.66(m,6H),7.73-7.8(m,3H);13 13 C NMR(125MHz,CDCl3 ):d36.00,120.63,122.47,125.03, 133.21, 137.43, 138.14; MS (EI, 70 eV, %): m/z 582(M , 10);元素分析计算值:C,55.42;H,3.62;实测值C,55.36;H,3.55。
2.2.3. 1,3,5-三(苯甲基三乙基溴)苯(TBTABB)的合成
将3g(5.12mmol)1,3,5-三[4-溴甲基苯基]苯,1.81g(17.92mmol)三乙胺和70ml乙腈的混合物置于150ml三颈圆头中。耐热玻璃烧瓶。使用配备有聚(四氟乙烯)(PTFE)半月形叶片的机械搅拌器连续搅拌反应混合物,所述半月形叶片在氮气氛下以600rpm搅拌。反应在70℃下进行24小时并轻轻回流。然后在真空下完全除去溶剂,用正己烷(3times;10ml)洗涤鎓盐5。将黄色固体MPTC储存在CaCl 2 干燥器中。熔点:62℃;收率:89%;红外 (kbr), (cm-1 : 1008, 1452,1688, 2963, 3357;1 1 H NMR(400MHz,CDCl3 ):d 1.37-1.41(t,27H,-CH 3 ),3.20(q,18H,-CH 2 ),3.45(s,6H,-CH 2 ),7.59-7.95(m,15H,ArH);13 c 核磁共振 (100 兆赫, CDCl3 : d 8.37、52.83、60.48、126.54、127.38, 127.76, 133.16, 140.93, 141.42;元素分析:计算。C,60.81;H, 7.49;N, 4.73;实测值C,60.75;H,7.44;N,4.69。
3. 两相反应的动力学
动力学实验在普通的光滑壁150ml三颈烧瓶中进行,该烧瓶配有平叶搅拌桨和回流冷凝器。通过反向添加方法进行反应[28].在三颈烧瓶中,将茚(1ml,8.52mmol),20%NaOH水溶液(w / v),即20g NaOH溶于100ml水(w / v,25ml),十六烷中的混合物(取出1.34g)和催化剂(5.6times;10-5 mmol)并在60℃下以200rpm搅拌5分钟以调节催化剂。将搅拌速度增加至500rpm,然后在零时间加入20ml 1,4-二溴丁烷。在停止搅拌过程时几乎立即发生相分离。从混合物的有机层收集样品(通过每次停止搅拌10-15秒),以规则的时间间隔。将少量无水CaCl 2 置于样品瓶中以吸收有机层中存在的任何水分。每次运行包括在5至30分钟的时间段内采集的七个样品。动力学之后是使用气相色谱仪估计消失的茚的量。
色谱柱(5%SE-30 Chrom WHP 80 / 100,3 mx 1/8英寸不锈钢填充柱)保持在200℃。一份反应混合物(0.5毫升)注入并获得1,4-二溴丁烷(1.09分钟),茚(0.95分钟),螺[环戊烷-1,1-茚](螺环衍生物)(2.01分钟)和十六烷(3.3分钟)的保留时间。向冰冷的反应混合物中加入乙醚(20ml),用水反复洗涤醚层,用无水硫酸镁干燥。使用硅胶柱色谱法纯化化合物。对于预期的产物,螺[环戊烷-1,1-茚]和1-(4-溴丁基)-1-茚,光谱数据与这些数据一致。光谱数据:FT-IR,(KBr),cm-1 :700,750,1000,1437,1600,1738,2200, 2975;
1 1 H NMR(400MHz,CDCl3 ):d 1.52-2.72(m,8H,-CH 2 ),6.69(d,1H,J = 5.36,-CH),6.85(d,1H,J = 5.36,-CH),7.28-7.55(m,4H,芳香族);13 cnmr (100 mhz, CDCl3 : d 26.01, 35.50, 60.56,120.86, 121.30, 124.98, 126.27, 127.62, 143.27, 144.97, 152.40.
4. 结果和讨论
在成功合成了一种新的多位点相转移催化剂MPTC后,我们通过选择茚6与1,4-二溴丁烷7的环戊烷作为模型反应来确定控制MPTC催化活性的几个实验参数的影响(方案2).动力学实验在假一级条件下进行,过量的氢氧化钠水溶液和1,4-二溴丁烷,得到环化产物8.C-烷基化的动力学然后估算使用气相色谱法消失的茚的量。从log(a-x)对时间的曲线图评估伪一阶常数,其中(a-x)是任何时间(t)的茚浓度。
4.1. 搅拌速度的影响
发生反应的必要条件是反应物分子的有效碰撞,甚至在相转移催化体系中也是如此。为了确定搅拌速度对反应速率的影响,我们在MPTC作为催化剂存在下,在50℃下在假一级反应条件下将搅拌速度从200转/分钟改变为800转/分(图1).从log(a-x)对时间的线性图评估伪一阶速率常数。结果表明,随着搅拌速度从100rpm增加到500rpm,反应速率线性增加。这是因为每单位体积分散体的界面面积随着搅拌速度的增加而线性增加,直至达到500rpm,其中每单位体积分散体的界面面积没有显着增加,相应的速度增加。因此,提高搅拌速度会改变分散相的粒径。因此,为了研究茚的C-烷基化的其他动力学变量,我们将搅拌速度保持在500rpm。高于500rpm,粒径没有显着变化。在高搅拌速度,即600-800rpm下,反应速率常数k降低。这可能不是由于茚浓度的降低。这可能是由于反应物(茚)与烷基化剂(1,4-二溴丁烷)和催化剂(TBTABB)的不适当碰撞,导致它们之间的接触时间减少。此外,传质已达到恒定值。因此,图。1表明界面反应机制而不是斯塔克斯提取机制。在两相反应系统中,搅拌速度对反应速率的影响可以通过(i)斯塔克斯的萃取机理和(ii)Makosza的界面反应机理来解释。对搅拌速度变化影响的动力学研究揭示了界面机制[29–31] 转移速率限制低于给定的搅拌速度(600-800rpm)并且固有反应速率限制在该搅拌速度以上。涉及提取机制的反应,由Starks和Owens提出[32],显示类似的性能,但在物理和化学控制(100-300转/分钟)之间的搅拌速度的限制较小。Chiellini等[33] 据报道,即使搅拌速度达到1950转/分钟,PAN的乙基化速率也不断提高;他们提出了一种界面机制。Murugan和Gopinath[21] 观察到速率常数对搅拌速度的线性依赖性,使用新的鎓盐对吡咯的N-烷基化进行高达500rpm的转化,其中提出了界面机理。
4.2. 不同底物量的影响
茚的浓度从4.26变化到在保持1,4-二溴丁烷和15%(w / v)
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[415788],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 通过对奥美拉唑合成反应的监测和定量反应的在线拉曼光谱和表征组件外文翻译资料
- 无金属碳基催化剂的研究进展外文翻译资料
- 钼酸钙/碳三维复合材料可控设计合成的研究外文翻译资料
- 生物催化选择性合成功能化喹唑啉酮衍生物外文翻译资料
- 三元V Zr Al ON氧氮化物-3-甲基吡啶氨氧化的高效催化剂外文翻译资料
- 综述纳米零价铁(nZVI)的合成,特性和在环境修复中的应用外文翻译资料
- 自消毒PVC表面使用点击化学设计外文翻译资料
- 微波辅助直接合成4H-1,2,4-苯并噻二嗪1,1-二氧化物衍生品外文翻译资料
- 微波辅助下直接合成1,1-二氧代-4H-1,2,4-苯并噻二嗪类衍生物外文翻译资料
- 压力选择在变压精馏中的重要性外文翻译资料

