N-杂芳烃的自由基氧化C-H烷基化反应的研究进展
原文作者:Jun-Rong Zhang, Li Xu, Yan-Yan Liao, Jian-Chao Deng, and Ri-Yuan Tang*
单位:Department of Applied Chemistry, College of Materials and Energy, South China Agricultural University, Guangzhou, Guangdong 510642, China
摘要:本文综述了N-杂芳烃通过自由基氧化偶联过程进行碳氢烷基化的研究进展。综述了用于N-杂芳烃的各种烷基自由基来源(包括烷烃、醚、醇、羧酸、有机烷和其他烷烃衍生物)。
关键词:自由基;氧化偶联;C-H功能化;烷基化;杂芳烃
介绍
自由基氧化C-H功能化已成为C-C和C-杂原子键形成的有力工具[1]。与传统的耦合方案相比,自由基氧化C-H功能化具有阶梯经济、原子经济、高效、操作简单、反应条件温和等优点。它不需要引导基团或重金属催化剂,而这些在过渡金属催化的C-H活化中往往是必不可少的[2]。因此,N-杂芳烃的自由基氧化C-H烷基化反应得到了很大的关注(方案1)[3-5]。自由基氧化C-H烷基化可以追溯到1971年报道的Minisci反应[3]。这项开创性的工作表明,在过硫酸铵存在下,由新戊酸脱羧生成的丁基自由基可以与质子化的吡啶偶联,实现吡啶的C-H烷基化。1982年,Tingoli和合著者[4]报道了二叔丁基乙酯(过氧化物酸盐)可以诱导与未质子化的吡啶结合的二氧烷基和环己基自由基的生成。受这些早期工作的启发,人们对自由基氧化C-H转化进行了广泛的研究。例如:CDC(交叉脱氢偶联)反应,[6]光氧化还原碳氢转化[7]。
众所周知,N-杂芳烃广泛存在于生物系统、药物、材料、农用化学品和摄影中。N-杂芳烃的自由基氧化C-H烷基化已经成为一个日益发展的领域,因为它为纳入结构多样性提供了更多的选择。在大多数情况下,C-H烷基化需要预先功能化的N-杂环芳烃,如N-氧化物和N-亚胺吡啶[8]。因此,直接烷基化的非活化的N-杂芳烃在成本和简单性方面非常有吸引力。近年来,各种烷基自由基前置剂(包括烷烃、醚、醇、醛、羧酸、氨基酸和其他烷烃衍生物)被用于N-杂芳烃的C-H烷基化。本文综述了不同烷基源的N-杂芳烃C-H烷基化的研究进展。
方案1 N-杂芳烃的自由基氧化碳氢烷基化反应
讨论
烷烃作为烷基自由基源
烷烃生成的烷基自由基通常由有机过氧化物引发,如TBHP(叔丁基过氧化氢)和DTBP(二叔丁基过氧化氢)[4,6]。在某些情况下,需要金属催化剂来促进偶联反应。例如,Li组[9]发现Sc(OTf)3在DTBP的存在下,能有效地促进吡啶衍生物与环烷烃的烷基化作用(方案2)。吡啶的缺电子性质导致低反应性,这需要Lewis酸如Sc(OTf)3以提高吡啶上C(2)-H键的酸度。在Sc(OTf)3的帮助下,几种环烷烃与吡啶衍生物反应顺利,得到烷基化产物,产率为51%-91%。在大多数情况下,在反应中得到了单烷基化产物和二烷基化产物的混合物。
方案2
嘌呤核苷也可以作为烷基自由基的有效受体。嘌呤核苷的C-H环烷基化在140 ℃的金属自由基下进行(方案3和4)[10,11]。嘌呤上的游离氨基对140 ℃处的DTBP具有良好的耐受性(方案3)。有趣的是,当C(6)和C(8)-反应位点同时存在时,C(6)-单环烷基化C(6)-或C(8)-二环烷基化C(6)-可以通过控制DTBP负载和反应时间选择性地获得嘌呤核苷(方案4)。该方案还允许尿嘧啶和相关核苷具有良好的区域选择性。
方案3
方案4
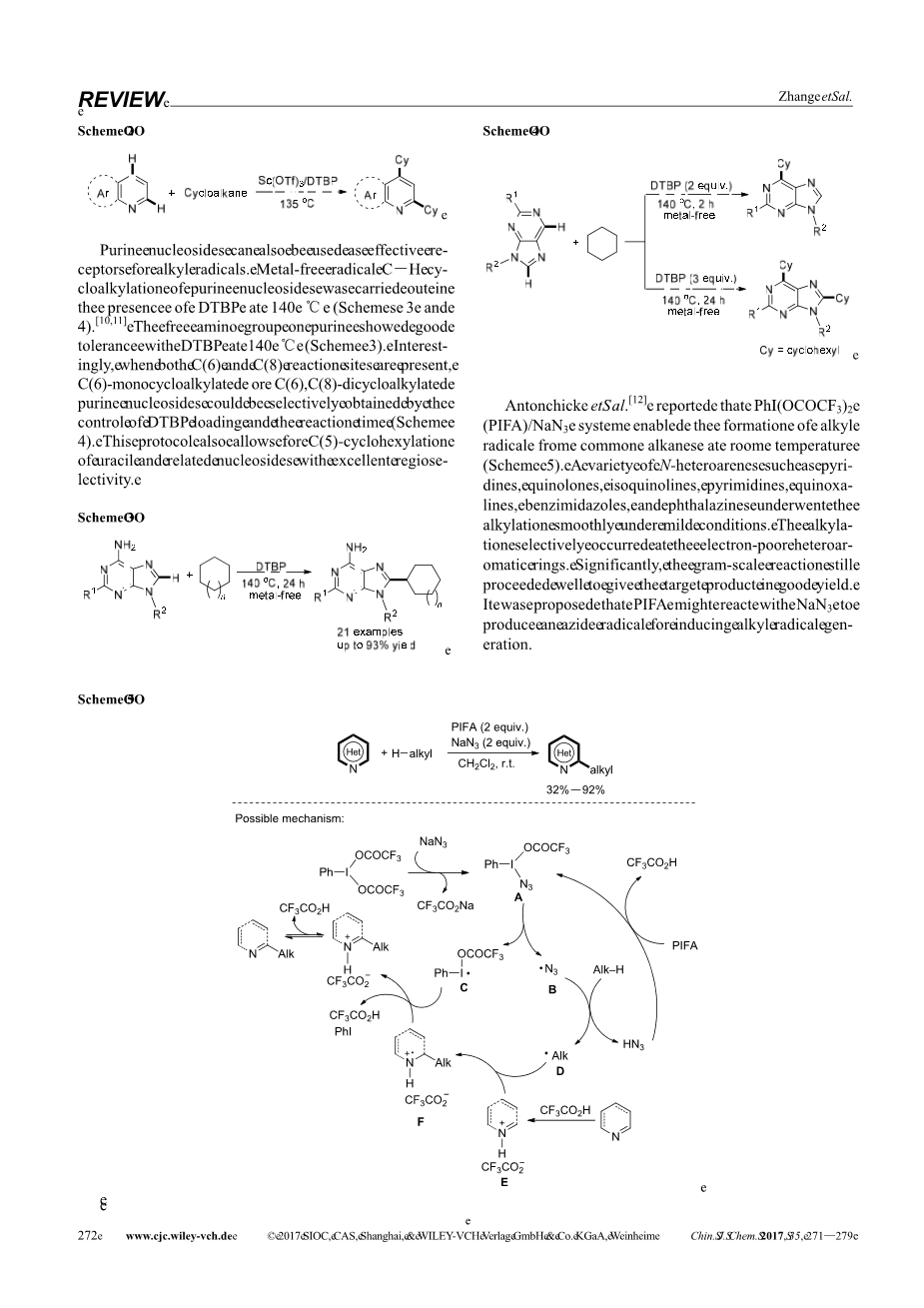
Antonchick等人[12],报道PhI(OCOCF3)2(PIFA)/NaN3系统能在室温下由普通烷烃生成烷基自由基(方案5)。各种N-杂芳烃,如吡啶、喹诺酮类、异喹啉类、嘧啶、喹恶啉类、苯并咪唑和苯酞嗪类在温和条件下顺利进行烷基化。烷基化选择性地发生在缺乏电子的杂芳环上。值得注意的是,克级反应仍然进行良好,得到了良好的收率。有人提出PIFA可能与NaN3发生反应生成用于诱导烷基自由基生成的叠氮化物自由基。
方案5
醚类/醇类作为烷基自由基来源
烷基醚和醇都是C-H烷基化的常见自由基前体[13]。自由基中心的产生通常发生在醚或醇的氧原子附近的碳氢键上。其中,有机过氧化物和过硫酸盐被广泛用作自由基氧化偶联反应的有效烷基自由基引发剂。2011年,Wang等人[14],证明了在没有金属催化剂的情况下,在120 ℃的TBHP存在下,可以通过自由基氧化偶联过程与醇或醚进行直接C(2)-烷基化(方案6)。唑类物质包括苯并噻唑、苯并恶唑和苯并咪唑等,都适用于这种烷基化反应。伯醇和仲醇均表现良好,可提供中等到良好产量的羟基烷基化产物。
方案6
与有机过氧化物相比,过硫酸盐更稳定,因为有机过氧化物在高温反应中更容易处理。因此,在温和的条件下,由过硫酸盐诱导的C-H烷基化是非常可取的。Jiang等人[15]报道了一种铜催化的(苯并)噻唑与环醚的C-H烷基化反应,使用K2S2O8作为氧化剂(方案7)。有趣的是,该方案也适用于非苯并融合的唑和非活性醚之间的烷基化。
方案7
Singh等报道了K2S2O8在不使用任何金属催化剂的情况下,可以在120 ℃下在丙酮和H2O(V:V = 2 : 1)的混合物中引发环醚与 N-杂环芳烃的烷基化反应(方案8)。多种缺电子杂芳烃(如非取代异喹诺酮类、喹啉类、吡啶、吡嗪和嘧啶类)适合烷基化。此外,本方法也适用于不含三氟乙酸情况下的缺电子萘醌[17]。这种无金属的C-H烷基化策略可能对药物合成很有吸引力,因为它可以防止药物的金属污染。
为了在温和的条件下实现C-H烷基化,光氧化还原策略有望在室温下实现C-H烷基化。MacMillan组[18]在室温下,开发了一种可见光促进的缺电子杂芳烃与醚的直接alpha;-氧烷基化反应(方案9)。这种温和的方案允许即使是高挥发性的乙醚如乙醚偶联,并允许使用原料底物和商业光催化剂直接获得广泛用途的药用药物载体([Ir{dF(CF3)-ppy}2(dtbbpy)]PF6)。
方案8
方案9
在自由基氧化C-H烷基化反应中,富电子的吲哚作为自由基受体仍然存在。最近,Kwong等人[19]发现在无金属条件下,带有吸电子基团的吲哚溶液可以进行自由基氧化C-H烷基化(方案10)。在DTBP的帮助下,许多非活性环醚和环烷烃直接与吲哚衍生物偶联,产率令人满意。对溴、酮、丁腈、酯、醛等吸电子基团的耐受性良好。但在C(2)或C(3)位置有供电子取代基的吲哚没有讨论。
方案10
通过自由基氧化C-H烷基化过程,还可以讲羟烷基团附着到杂环芳烃上。Neubert等人[20]在TBHP和TiCl3的存在下,开发了3,6-二氯吡啶类化合物与不同烷基醇的羟基烷基化反应(方案11)。该反应是在空气中进行的,在克尺度上,没有产率变化。值得注意的是,具有氨基的烷基醇也适合烷基化反应,羟基烷基化产物可以与胺偶联环化,产生具有多官能团的新型融合吡啶类。
方案11
早期有报道称,在高能照射下,醇可以通过碳氧键断开产生烷基自由基。 [21]受到这些作品的启发,MacMillan小组[22]在室温下,通过光氧化和硫醇氢原子转移有机催化的协同作用,开发了N-杂芳烃与醇的C-H烷基化的光氧化策略(方案12)。多种N-杂芳烃,如异喹啉、喹啉、酞嗪、苯嗪、菲啶和吡啶类,以甲醇作为甲基自由基源顺利进行甲基化。在这种双催化方案中,各种醇可作为有效的烷基化剂。这种温和的烷基化过程是一种强大而通用的方法,使用商业上可用的和丰富的醇作为潜在的烷基化剂。
Lectka组[23]发现,在二氧化锰(IV)和TFA的存在下,环丙醇开环反应,直接烷基化提供多种含酮的烷基化杂芳烯,具有广泛的官能基团耐受性。所提出的反应机制表明,锰的两种不同的氧化态(III和IV)可能在环丙醇C-C键的裂解和再芳构化步骤中发挥作用(方案13)。
方案12
醛和羧酸作为烷基自由基来源
据报道,脂肪族醛可以为缺电子杂环的烷基化提供一种烷基自由基。[24]通过过氧苯甲酸叔丁酯(TBP)处理,脂肪族醛在没有金属催化剂的情况下进行脱碳化过程,生成烷基自由基,然后与各种缺电子杂芳烃偶联,在130 ℃下产生烷基化的N-杂芳烃12小时(方案14)。
方案13
方案14
有机过氧化物并不总是由醛生成烷基自由基所必需的。例如,在氧的存在下,烷基醛的脱碳化仍然可以形成烷基自由基(方案15)[25]。1.5当量的TFA对N-杂芳烃的质子化作用对提高反应活性至关重要。乙基、异丙基、叔丁基、环己基醛均表现良好,产率为40%-90%。多种N-杂芳烃都适用于烷基化反应,即当N-杂芳烃上存在多个反应位点时,就得到了单烷基化产物和双烷基化产物(或三烷基化产物)的混合物。所提出的反应机理表明,醛的自氧化会产生酰基自由基A,然后在加热条件下传递相应的烷基自由基,进行N-杂芳烃的烷基化。
近年来,以脂肪酸为烷基自由基前驱体,以CO2为唯一副产物的微型反应得到了广泛的关注。Guo等[26]人在室温下与嘌呤核苷脱羧烷基化反应(方案16)进行了区域选择性极小反应,该反应对两种嘌呤核苷(如核糖核酸、脱氧核糖、阿拉伯嘌呤核苷)和脂肪酸(包括初级、二级和三级脂肪族羧酸)均具有良好的耐受性。含氯基和三氟甲基基的含电子缺陷嘧啶也适用于微型反应(方案17)[27]。各种现成的烷基和(异)环烷基羧酸的烷基化反应良好,产率较好。
烷基硼烷作为烷基自由基源
据报道,有机硼烷是通过氧化碳-硼键裂解形成C-C键的有用的自由基前驱体[28]。近年来,烷基硼烷被用于微小反应。Molander组[29,30]首先利用烷基兰烷氧基钾-甲基三氟硼酸盐作为N-杂芳烃烷基化的亲核自由基前驱体(方案18)。Mn(OAc)3被发现对从有机硼烷中生成烷基自由基有效。该方案是一种引入独特的烷基取代基的有效方法。(例如环丁基和烷氧甲基)转移到不同的N-杂芳烃(包括喹诺酮类、异喹啉、苯并嘧啶、苯并咪唑和苯并噻唑)。
方案15
方案16
方案17
方案18
Chen[31]组采用烷基硼酸作为自由基前体,通过光氧化还原策略进行N-杂芳烃的C-H烷基化反应(方案19)。以[Ru(bpy)3]Cl2为光催化剂和乙酰氧基苯并咪唑(BI-OAc)为催化剂,在温和条件下,广泛的一级和二级烷基有效地与各种N-杂环芳烃相互作用。该反应表现出前为药物分子的后期功能化提供了一种广泛适用的方法。在烷基化反应中,由光激发的钌(Ⅱ)的SET引发的活性物种BI-OAc对于烷基硼酸形成烷基自由基至关重要。
方案19
其他烷基自由基来源
DiRoccol和共同作者[32]报道了一种可见光光氧化还原催化诱导有机过氧化物产生烷基自由基,使具有重要药物意义的先导物和候选药物的后期功能化(方案20)。所描述的独特的温和的条件为了生成甲基和环丙基自由基,允许有效地捕获各种具有生物活性的N-杂
剩余内容已隐藏,支付完成后下载完整资料

英语原文共 9 页,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[597694],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 通过对奥美拉唑合成反应的监测和定量反应的在线拉曼光谱和表征组件外文翻译资料
- 无金属碳基催化剂的研究进展外文翻译资料
- 钼酸钙/碳三维复合材料可控设计合成的研究外文翻译资料
- 生物催化选择性合成功能化喹唑啉酮衍生物外文翻译资料
- 三元V Zr Al ON氧氮化物-3-甲基吡啶氨氧化的高效催化剂外文翻译资料
- 综述纳米零价铁(nZVI)的合成,特性和在环境修复中的应用外文翻译资料
- 自消毒PVC表面使用点击化学设计外文翻译资料
- 微波辅助直接合成4H-1,2,4-苯并噻二嗪1,1-二氧化物衍生品外文翻译资料
- 微波辅助下直接合成1,1-二氧代-4H-1,2,4-苯并噻二嗪类衍生物外文翻译资料
- 压力选择在变压精馏中的重要性外文翻译资料

