
英语原文共 6 页,剩余内容已隐藏,支付完成后下载完整资料
微结构表面统一可控厚度的共形纳米涂层的普适性组装策略
人们长期致力于研究在各种表面装载纳米粒子,用于构建功能性纳米涂层。然而,在微结构或纳米结构表面构建均匀的纳米涂层依然是一个挑战。本文证明了带负电的SiO2纳米粒子和带正电的硅偶联剂可以通过静电作用和缩合反应逐层的组装到微结构表面。具有可控厚度的纳米涂层构建在不同组成和不同形貌的微结构表面。利用QCM-D研究实时装配过程确定形成原理。通过研究具有相反电荷的其他活性反应物质形成共形纳米涂层的过程,证明了这种方法的普适性。非平面光学材料上的增透性纳米涂层的制备已经有应用。这种简单、通用、可扩展性的构建共形纳米涂层的方法在实际应用方面有很好的前景。
微结构表面有许多重要应用,微结构表面的功能纳米涂层有很高的价值。例如,菲涅尔透镜、电视屏幕等非平面光学设备上的增透纳米涂层可以提高透明度、亮度和对比度。
对于平面表面,人们研究出了许多构建可控厚度的功能纳米涂层的方法,例如自组装,旋转涂布,浸涂法以及化学气相沉积法。但是在微结构表面构建共形,可控厚度的纳米涂层更复杂。最近,人们研究了区域选择性原子层沉积法来实现薄膜的选择性生长,其具有很好的正形性,原子厚度可控性和大面积均匀性。但是这项技术有复杂的步骤,需要昂贵的仪器,而且只适用于薄膜上纳米结构的构建。
从微结构表面的分散体组装纳米粒子,从而构建共形纳米涂层更加方便。然而分散体的重力、表面张力、表面弹力会引起纳米粒子在微结构表面的不均匀分散,且纳米粒子干燥过程中,其间的毛细作用力也会引起颗粒团聚。这些因素均会导致合成出的纳米涂层有缺陷或不均匀。
本文阐述了用三氨丙基三乙氧基硅烷和二氧化硅纳米粒子作为骨架,可以在微结构表面很容易的构建共形纳米涂层。通过静电作用和缩合反应,带异性电荷的活跃骨架粒子可以逐层的均匀形成在微结构上,同时其间的多种相互作用可以在干燥过程中维持稳定。纳米涂层的厚度可通过反应时间来控制。这种方法可运用于从菲奈尔透镜上的微米尺度到蝉翼上的纳米棒,其他带相反电荷的活跃粒子也适用,证明这种方法是具有普适性的。这种简易、普适、可扩展的方法在功能性纳米涂层和非平面基片的实际应用中有很好的前景。
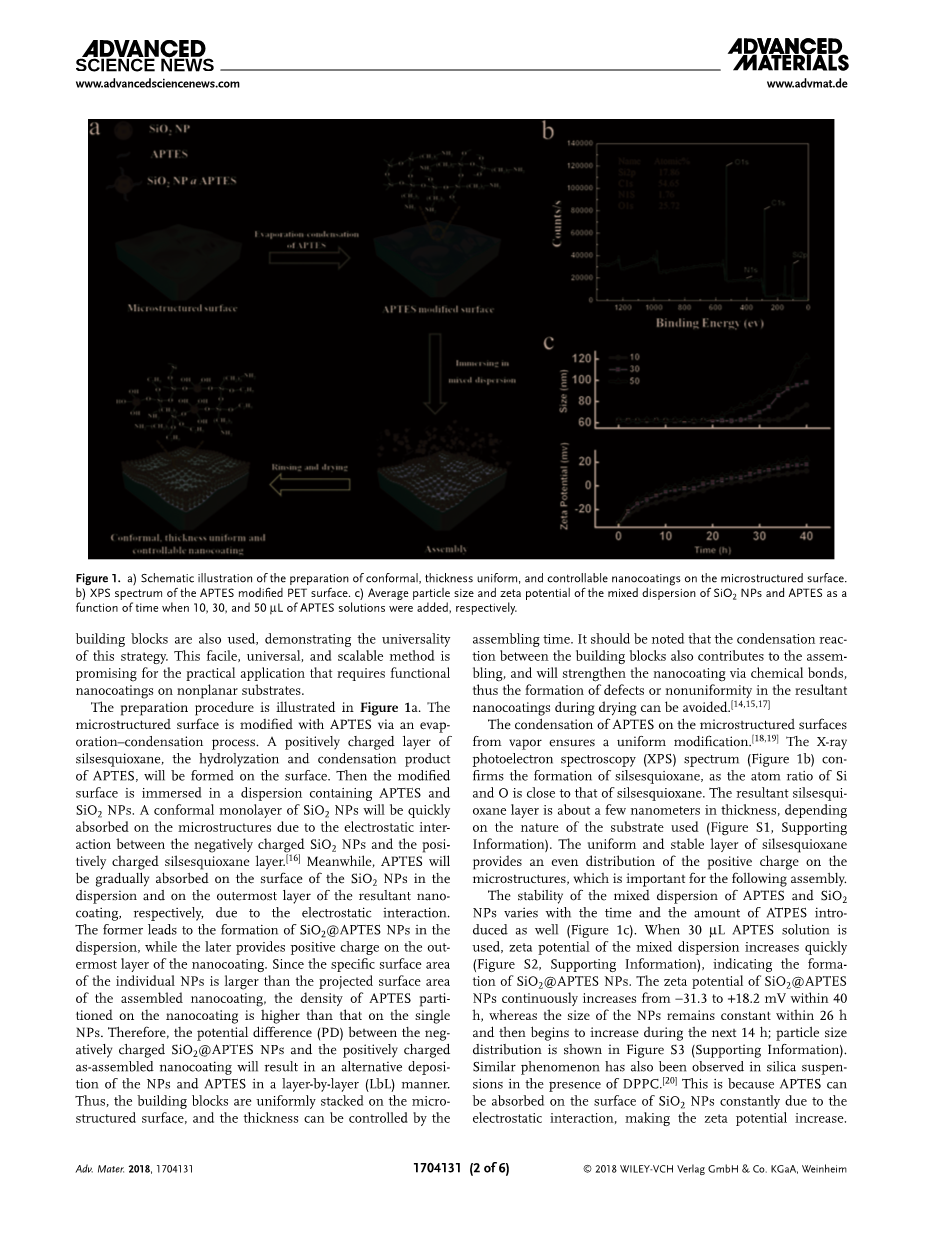
制备过程如图1a。通过蒸发冷凝将APTES修饰在微结构表面,APTES水解和缩聚后,在表面形成一层带正电荷的倍半硅氧烷。然后将修饰过的微结构表面浸泡在含有APTES和二氧化硅纳米粒子的溶液中,通过带负电荷的SiO2纳米粒子和带正电荷的倍半硅氧烷之间的静电作用,微结构表面会迅速形成单层共形的SiO2纳米粒子。同时,APTES通过静电作用会被逐渐吸附到分散层和纳米涂层的最外层的表面上。
前者形成分散液中的SiO2@APTES纳米粒子,后者形成带最外层正电荷的纳米涂层。由于单个NPs的表面积大于已组装的纳米涂层的表面积,因此在纳米涂层上的APTES的密度比单个NPs的密度要高。因此,带负电荷的SiO2@APTES NPs和带正电荷的纳米涂层之间的电位差(PD)将导致NPs和APTES产生替代沉积。因此,在微结构的表面上,所有的构件都是均匀堆积的,厚度可以由组装时间来控制。需要注意的是,反应物之间的缩合反应也有助于组装,并通过化学键加强纳米涂层,从而避免了在干燥过程中形成的纳米涂层的缺陷或不均匀性。
在微结构表面上的apte的冷凝可以保证均匀改性。x射线光电子能谱(XPS)光谱(图1b)证实了硅氧烷的形成,因为硅和氧的原子比接近于硅氧烷。合成的硅氧烷层厚度约为几纳米,这取决于所使用的基板的性质(图S1)。均匀和稳定硅氧烷层提供了微结构上的均匀的正电荷分布,这对下面的组装很重要。
apte和SiO2 NPs混合分散相的稳定性随时间和所引入的atpe的数量而变化(图1c)。当使用30 micro;L apte溶液时,混合分散相的界面动电势会迅速增加(图S2),表明SiO2@APTES NPs的形成。SiO2@APTES NPs的界面电动势在40小时内从-31.3增加到 18.2 mV,而NPs的大小在26小时内保持不变,然后在接下来的14小时内开始增加;粒度分布见图S3。在DPPC存在的硅悬浮物中也发现了类似的现象。这是因为由于静电的相互作用,apte可以持续的被吸收到二氧化硅纳米粒子表面,从而使界面电动势增加。apte壳体的缩合反应导致了聚合,导致在接下来的14个小时内粒子尺寸的增加。透射电子显微镜(TEM)图像显示纳米粒子表面的粗糙度随时间增大。然后分散相就变得不稳定了。在第60个小时时可以观察到NPs的沉积,在TEM图像(图S4e,支持信息)中出现了20-30纳米的新聚合体,这可以归因于apte的水解和缩合。在我们的例子中,稳定和带负电荷的SiO2@APTES NPs(在20个h以内)作为组装的构建块。
QCM-D在现场实时监控组装过程。当晶体晶片的质量随着吸附而增加时,共振频率会降低,而能量耗散参数则提供了吸附层粘弹性特性的信息。图2a显示,在混合分散相被注入到腔室后,共振频率急剧下降。相应的扫描电镜图像显示了沉积在芯片上的单层NPs(图2),表明带正电的芯片可以快速吸附一层带负电荷的NPs层。在此之后,Delta;f 逐渐减少,NPs继续在芯片表面组装。SEM图像显示得到了厚度为360plusmn;10nm的均匀纳米涂层(图2j)。在用乙醇清洗的过程中,没有观察到Delta;f的明显变化,这表明已经形成了一种稳定的纳米涂层。在装配过程中,Delta;D会相应地增加。qcm-d的结果表明,装配过程可以分为两个阶段:第一阶段的快速沉积阶段,第二阶段是逐步组装的阶段。
装配的驱动力主要是在分散和表面上的构件之间的PD(图2d)。在一开始,高PD提供了一个强大的静电吸引,并有助于在第I阶段快速形成一个单层的NPs(图2)。这种现象在其他地方也得到了观察。由于对apte的持续吸收,分散相中SiO2@APTES NPs的界面电动势逐渐增加。因此,在合成的纳米涂层上的最外层的apte吸收能力就会降低。因此,在早期装配过程中,晶片表面的界面电动势可能会随着时间的推移而减小。逐渐减少的PD导致了在第二阶段的构建块的缓慢组装。在8小时后,SiO2@APTES NPs的界面电动势接近0,SiO2@APTES 纳米粒子吸收ATPES的能力下降。由于SiO2@APTES NPs和APTES之间的静电相互作用和缩合反应,在合成纳米涂层的最外层,apte的连续吸附会增加表面的界面电动势。在14小时后,SiO2@APTES NPs被带正电,并且几乎停止了组装。合成的纳米涂层上最外层的吸附平衡使表面势形成的纳米涂层平整。XPS结果在4和15 h,分别清晰地显示了增加的倍半硅氧烷,再一次证明了基于APTES的假设。当SiO2@APTES NPs被带正电时,装配可能会受到静电排斥的阻碍,而观察到的NPs的相对缓慢的沉积过程则可以归因于NPs和组装的纳米涂层之间的缩合反应。
为了进一步研究这一机制,进行了两项控制实验。当使用SiO2 NPs分散相时(图2b),在几分钟内也会获得同样的△f变化(图2b)。SEM图像还显示了芯片上单层NPs的形成(图2h)。但是,△f和涂层的厚度在接下来的20小时(图2b,k)中没有改变,并且表面的界面电位呈负电(表S1,支持信息)。结果表明,在分散相中没有apte的存在,SiO2 NPs不能连续地在表面上进行组装。此外,当apte溶液在第二阶段注射时,qcm-d结果(图2e)清楚地显示了在带负电荷的单层SiO2上的apte的快速吸收。当再次注入SiO2色散时,可以观察到类似于图2a的装配过程,进一步证明APTES是维持一定的PD作为装配驱动的必要条件。
在另一种情况下,当使用未改性的黄金芯片(图2c,c)时,△f的急剧减少不会出现。SEM图像(图2i)显示一些NPs在晶片表面上沉积,表明未修改的晶片不能吸附带负电荷的NPs。然而,胺基可以在溶液中通过14.2 and 38.5 kJ molminus;1的结合能与黄金结合。因此,NPs可以逐渐在芯片上装配。顶部和横截面扫描图(图2l)显示了非均匀纳米涂层的形成。如果使用没有与硅氧NPs或APTES相互作用的基质,如聚(乙烯基苯二酯)(PET)或硅晶片,SEM图像(图s6a-d,支持信息)显示纳米涂层不能在这样的表面形成。控制实验表明,在分散相中APTES的存在和APTES改性表面是形成均匀和可控制的纳米涂层的关键因素。
分散相中APTES量的不同会影响基板和NPs之间的PD,进而改变了装配的速率(图7,支持信息)和合成纳米涂层的厚度(图2f)。PD的逐渐降低确保了第二阶段的组装能够逐层进行(图8),由于PDn比PDn 1大,第n层和NPs之间的静电吸引大于(n 1)层和NPs之间的静电吸引,使得大多数NPs更倾向在第n层上叠加。尽管逐渐减少的PD导致了装配速度的降低,但我们的在一个容器中的装配方法比传统的在不同溶液中的装配方法的优点多。
图3显示了在表面形成的共形纳米涂层的SEM图像,微结构有不同的成分和形态。例如,在PET褶皱的侧壁或顶部可以获得大约150纳米的纳米涂层。与之相反,在同一基体上涂覆的二氧化硅NPs在顶部上沉积的很薄,而合成的纳米涂层的厚度从顶部到底部沿侧壁增加(图9,支持信息)。更有趣的是,即使是在蝉翼上的纳米足上,NPs也会均匀地覆盖在纳米棒表面,而不会破坏原始的阵列结构。
为了说明这一策略的普遍性,一个包含带负电荷的单宁酸(TA)和带正电荷的SiO2@APTES NPs的系统被应用在一个TA修改的芯片上(图S10,支持信息)。当添加TA时,SiO2@APTES NPs分散相的界面电动势从 8.3下降到 3.1 mV(图11,支持信息)。qcm-d结果显示了类似的组装行为(图S10c,支持信息)。经过20小时的改性后,纳米涂层的厚度为400plusmn;10纳米。当采用带负电的二氧化硅NPs分散相时,由于TA对SiO2 NPs的静电排斥,不会发生装配。
应该注意,粒子的大小和质量会影响组装,因为稳定的分散相是保证纳米涂层均匀性的前提条件,而静电的相互作用应该足够强,能够对抗NPs的引力的影响,使装配顺利进行。我们发现用220nm二氧化硅NPs和聚二甲基二烯酰铵也可以构建均匀的纳米涂层,它的正电荷密度更高(图S12,支持信息)。
由二氧化硅NPs制成的纳米涂层已被广泛应用于光学材料的AR涂层。对于单层的AR涂层,要实现全反射,最理想的厚度是,lambda;是入射光的波长,是涂层的折射率,, 和分别是空气和基板的折射率,是入射角。对于波长在400-800纳米的可见光范围内,d的范围是(100-200)nm/。在这个范围之外透光率会下降。因此,对厚度和均匀性的精确控制是实现最佳增透的必要条件。该方法的优良可控性,使其适于在非平面光学基板上制备AR纳米涂层。在此基础上,通过调整浸泡时间至6h小时、8小时和20小时,在微结构PET表面上得到了厚度为115plusmn;4、149plusmn;1和273plusmn;10纳米的纳米涂层。如图4a所示,厚度约为150纳米时透光率平均提高了2.6%,在最优厚度的范围内更有可能实现。相比之下,通过浸涂法(图15,支持信息)所形成的纳米涂层,透光率只提高了不到1%。采用这种方法,均匀的纳米涂层也在平菲奈尔透镜的同心圆上形成(图S16,支持信息),通过控制修改时间(图4b),可以达到最佳的增透性能。
概括地说,共形、厚度可控制的纳米涂层是构建在在微结构上的,尺寸在微米到纳米之间。纳米涂料的形成是活性和带相反电荷的构件之间的静电相互作用和化学反应共同作用的结果。该策略的可控性,特别是在纳米涂层的均匀性和厚度上,可以通过调整时间来达到亚波长范围。这一策略是非常通用的,对于在微结构的基板上制备增透纳米涂层方面具有许多优势。目前的方法有望提供一种简单而可扩展的方法,用于在各种微结构基板上进行纳米涂层的实际制备。
支持信息
来自威利在线图书馆或作者的支持信息。
声明
作者对中国国家自然科学基金(51522308,21474117,51673203)和科技部(2013CB933000,2015DFG32320)的财政支持表示感谢。作者感谢张庆松教授,
Xiaoxu Song教授和Xiaoxu Song对表面的潜在测量进行了帮助。作者们也非常感谢Xiaoyu Zhang和Xiaoyu Zhang对XPS的帮助。
关键词
增透涂料,装配,正形,微结构,纳米涂层
[1] J. J. Kim, J. Lee, S. P. Yang, H. G. Kim, H. S. Kweon, S. Yoo, K. H. Jeong, Nano Lett. 2016, 16, 2994.
[2] Y. W. Kwon, J. Park, T. Kim, S. H. Kang, H. Kim, J. Shin, S. Jeon, S. W. Hong, ACS Nano 2016, 10, 4609.
[3] P. Mazumder, Y. Jiang, D. Baker, A. Carrilero, D. Tulli, D. Infante, A. T. Hunt, V. Pruneri, Nano Lett. 2014, 14, 4677.
[4] P. M. Davidson, H. Ouml;zcelik, V. Hasirci, G. Reiter, K. Anselme, Adv. Mater. 2010, 21, 3586.
[5] L. Scandella, G. Binder, J. Gobrecht, J. C. Jansen, Adv. Mater. 1996, 8, 137.
[6] J. Choi, S. Song, M. T. Houml;rantner, H. J. Snaith, T. Park, ACS Nano 2016, 10, 6029.
[7] B. Thierry, F. M. Winnik, Y. Merhi, M. Tabrizian, J. Am. Chem. Soc. 2003, 125, 7494.
全文共7637字,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[10047],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 可调聚合物微球的简易合成及其在包裹色料中的应用研究外文翻译资料
- 非均质表面结构的金属有机框架用于癌症治疗,成像和生物传感的研究进展外文翻译资料
- 金属氧化物光阳极电荷产生到光催化的动力学综述外文翻译资料
- ILs基凝胶在储能、传感器和抗菌方面的研究进展外文翻译资料
- 水凝胶在水体污染物吸附和废水处理中的应用外文翻译资料
- 半互穿壳聚糖/离子液体聚合物网络作为伤口敷料和离子电渗透材料的应用研究外文翻译资料
- 碳纳米管/PLA复合材料的增材制造及构效关系外文翻译资料
- 单宁酸诱导环氧化大豆油交联增韧聚乳酸外文翻译资料
- 新型偶氮苯基两亲性共聚物:合成、自组装行为和多刺激响应特性外文翻译资料
- 用于多胺识别的光子晶体协同传感器芯片外文翻译资料

