
英语原文共 5 页,剩余内容已隐藏,支付完成后下载完整资料
扩大环丙烯离子相转移催化体系:苄基氟化
凯蒂·邓普西,罗亚·米尔,伊沃·斯马拉吉奇,特拉维斯·杜丁
关键词:环丙烯阳离子、氟化作用、相转移催化、氢键、苄基氟化
摘要:报道了环丙烯离子作为一种相转移催化剂在苯氟化反应中的应用。这些氟化反应机理的一个重要组成部分是原位衍生的氟化环丙烯配合物的作用,其存在得到 1H, 19F NMR和UV-Vis光谱的支持。通过密度泛函理论的计算,对这些反应的机理进行了深入的研究。
1.介绍
相转移催化(PTC)对化学合成的影响跨越了数十年,其在学术界和工业界的影响继续受到关注。1 造成这种状况的因素很多,包括操作简单,一般使用温和的反应条件和廉价的试剂/溶剂,以及可扩展性。催化剂介导的反应中间体在不同相(双相和/或三相)之间的迁移,进一步增加了其实用性,因为它减少了由不相容的化学反应性引起的副产物的形成。1,2
目前存在几类相转移催化剂,每种都具有化学反应性方面的明显优势。在这方面的经典是铵和鏻阳离子,其广泛用于在相之间穿梭阴离子物质。3,4 相反,冠醚通过主客体络合实现了阳离子的相转移。3,5 咪唑,6 三唑,7 和四氨基鏻8 离子也显示出作为相的效用,在这些情况下,具有作用模式的转移催化剂衍生自各种非共价相互作用,包括pp堆积,阳离子-p相互作用和H-键合。9,10 此外,我们小组最近率先使用环丙烯类似物作为相转移催化剂,11而在当代作品中,Lambert及其同事报道了使用三(二烷基氨基)环丙烯盐作为相转移催化剂的使用情况。12
就相转移催化应用而言,C-N,C-O和C-C键形成方案可以说是最成熟的,13 然而,C-F键的形成仍然没有被探索过。14 尽管氟化化合物在药物设计、农用化学品、材料科学、 18F正电子发射断层扫描成像(PET)和药物中具有价值。15 事实上,据估计,20-30%的药物分子含有至少一种氟,因此突出了开发制备有机氟化合物的方法的重要性。16 除此之外,氟化叔丁基铵(TBAF)已被广泛用于相转移催化的氟化反应,17 然而,与其使用相关的缺点是,例如,它是吸湿的, 经历Hoffman E2 消除发生在40和77 ℃(2托)之间,具有低氟化物占试剂总质量比例较低(F-=占NBu4F总质量的7.3%),在实践中,通常需要化学计量或更大量的总质量。此外,由于其强碱性,当使用无水TBAF时,经常观察到消除副产物。18 最后,水溶性产物(例如来自TBAF的糖)的分离可能具有挑战性,因为标准的后处理方案需要含水酸/碱洗涤。19
最近,利用相转移条件探讨了形成C-F键的替代方法。20例如,Toste和同事报告了一起使用手性磷酸盐在烯丙醇对映选择性氟化反应中难溶解的氟化试剂,21 2016年, Tamamura及其同事使用一种氟试剂,亲脂性阴离子相转移催化剂和碱的组合,开发了一种用于吡咯的温和氟化方法。22 此外, Hamashima最近开发了一系列二羧酸预催化剂,其在去质子化后用于烯烃不对称氟化的阴离子相转移催化剂。23
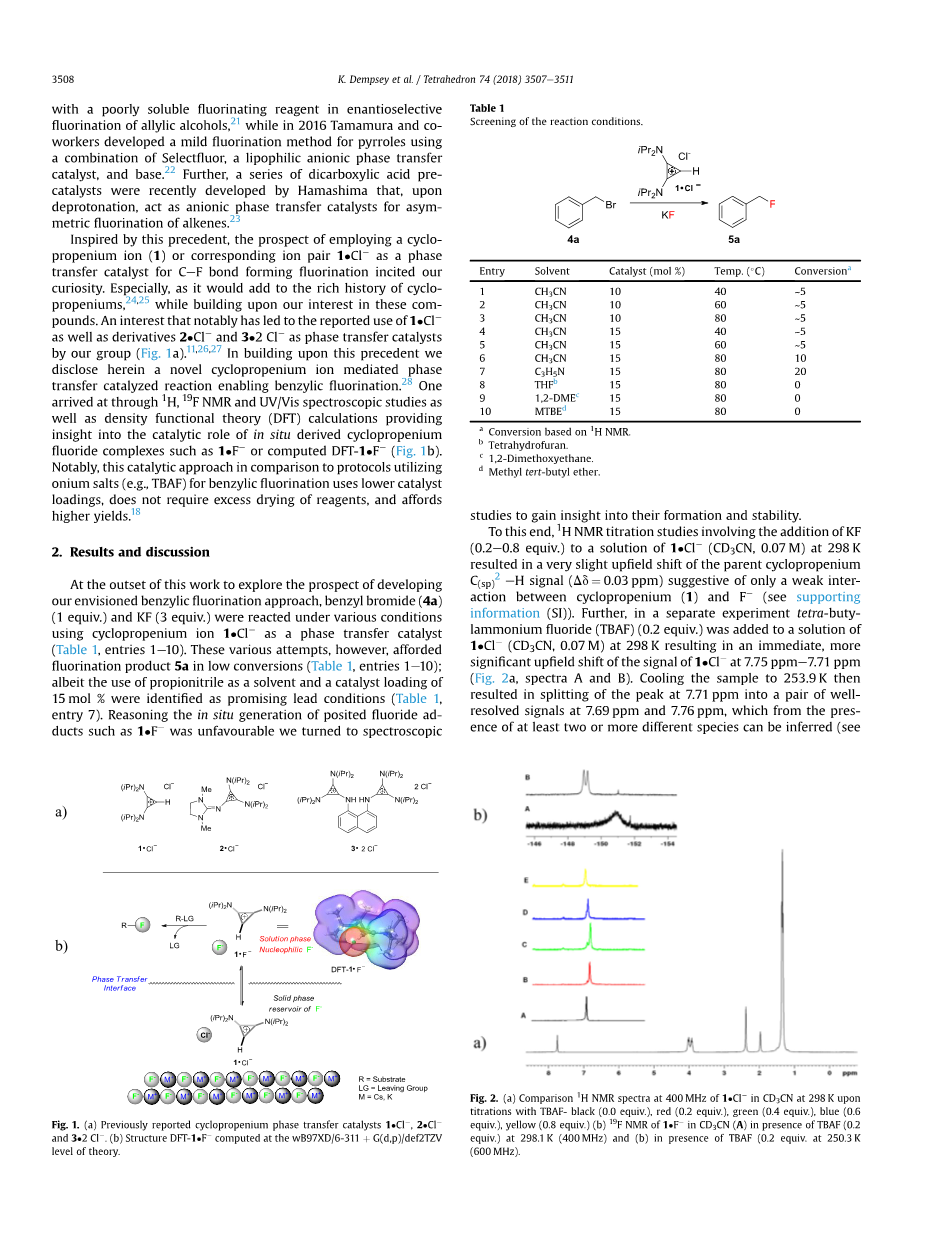
受这一先例的启发,采用环丙烯离子(1)或相应的离子对1·Cl-作为C-F键形成氟化的相转移的前景引起了人们的兴趣。特别是,这会增加环丙烯的丰富历史,24,25 同时建立在我们对这些化合物的兴趣上。我们小组对1·Cl- 以及衍生物2·Cl- 和3·2 Cl- 作为相转移催化剂的报道引起了极大的兴趣(图1a)。11,26,27 在此先例的基础上,我们在此揭示了一种新的环丙烯离子介导的相转移催化反应,使苯氟化。28 通过 1H, 19F NMR和UV/Vis光谱研究以及密度泛函理(DFT)计算得出的结果,提供了对原位衍生的氟化环丙基配合物如1·F- 的催化作用的见解,或计算DFT-1·F- (图1b)。值得注意的是,与利用鎓盐(例如,TBAF)进行苄基氟化的方案相比,这种催化方法使用较低的催化剂负载量,不需要过量干燥试剂,并且提供更高的产率。18
2.结果和讨论
在研究开发我们设想的苯氟化方法的前景时,使用环丙烯离子1 ·Cl- 在各种条件下使苄基溴(4a)(1当量)和KF(3当量)反应。相转移催化剂(表格1,条目1-10)。然而,这些不同的尝试提供了低转化率的氟化产物5a(表格1,条目1-10);虽然使用丙腈作为溶剂并且催化剂负载量为15 mol%被确定为有希望的铅条件(表格1,条目7)。推理原位产生假定的氟化物加合物如1bull;F- 是不利的,我们转向光谱研究以深入了解它们的形成和稳定性。
a基于 1H NMR的转化率。
b四氢呋喃。
c1,2-二甲氧基乙烷。
d甲基叔丁基醚。
为此,1H NMR滴定研究涉及在298K下向1·Cl-(CD3CN,0.07M)的溶液中加入KF(0.2-0.8 当量),得到了非常好的结果。导致母体环丙烯C(SP)2-H信号(0.03 ppm)的非常轻微的上场偏移,暗示环丙烯(1)和F-之间仅有微弱的相互作用( 参见支持信息(SI))。此外,在单独的实验中,将四丁基氟化铵(TBAF)(0.2当量)加入到1·Cl-(CD3CN,0.07M)的溶液中,在298K,产生立即的,更显著的高场1·Cl- 信号在7.75 ppme7.71 ppm处的偏移(图2a,光谱A和B)。将样品冷却至253.9 K,然后将峰值在7.71 ppm处分离成一对分辨率为7.69 ppm和7.76 ppm的信号,这些信号来自于可以推断出至少两种或更多种不同的物种(见SI)。同样,19F NMR显示室温下149ppm的宽信号(图2b,光谱A)在冷却至250.3K时,分解为149.20和149.98ppm处的两个峰(图2b,光谱B)。29 随后将样品升温至室温,然后加入另外的TBAF当量,然后导致环丙烯C(SP)2-H氢信号逐渐低场移位,为7.71 ppm—7.78 ppm(图2a,光谱C,D和E)。
图1.(a)先前报道的环丙烯基相转移催化剂1·Cl- ,2·Cl- 和3·2Cl- 。
(b)结构DFT- 1bull;F- 在wB97XD /6-311thorn;G(d,p)/ def2TZV理论水平下计算。
图2.(a)用TBAF-黑(0.0当量),红色(0.2当量)滴定,在CD3CN,298K下400℃下1bull;Cl- 的比较 1 H NMR光谱。在TBAF存在下,CD3CN(A)中的1bull;F- ,绿色(0.4当量),蓝色(0.6当量),黄色(0.8当量)(b) 19F NMR(0.2当量)在298.1K(400MHz)。
(b)存在TBAF(0.2当量,250.3K(600MHz))。
为进一步探讨1·F-配合物的形成,以TBAF(0.2当量)为例,对CH3CN中1·Cl-溶液的紫外可见光谱进行了监测。加入了这些加法的结果是 e在~325 nm处有一个明显的宽带出现,可能与非共价相互作用形成的1·F-,配合物有关(图3)。
图3. 5times;10-4M 1bull;Cl- 在乙腈中的溶液的UV-Vis滴定,其中0-2.0当量。TBAF。
有证据表明1·F-的形成复合物,并且仍然希望金属氟化物可用于该方法,由于它们的低成本和一般稳定性,我们将注意力转向使用CsF(740 kJ mol-1)相对于KF(821 kJ mol-1)的较低晶格能将促进盐复分解,导致形成1·F-。支持这一前提的是使用干燥CsF代替TBAF重复的NMR实验,这导致与用TBAF观察到的类似趋势(参见SI)。
根据这些观察,尝试用CsF(1.5mmol)和1·Cl-(15mol%)氟化苄基溴(4a)(0.5mmol),在87%的异醇中合成了苄基氟(5a)。总收益率(表2,条目1)。通过比较,在不存在催化剂的情况下观察到少于5%的产物(参见SI)。受这一有希望的结果的推动,进一步探索了该反应方法的底物范围。因此,具有吸电子硝基的4-硝基苄基溴(4b)在5小时内反应以提供5b,而在7小时后,2-(溴甲基)萘(4c)以84%的略低产率转化为产物5c(条目2和3)。另一方面,具有电感吸电子和共振给电子对氯取代基的基板4d 以89%的收率提供5d(条目4)。此外,给电子给予对甲基取代的4e 的反应得到产物5e,产率为92%,而将溴化物换成甲苯磺酸酯离去基团减弱了反应性(条目5和6)。同时,大体积的邻碘取代基4g的反应在10小时后产生5g产率降低81%,这可能是由于空间作用所预期的(条目7)。其次,在讨论区域选择性问题时,反应4h得到SN2位移产物5h,未观察到SN2rsquo;产物形成(条目8)。这种反应方法被证明是对初级底物有选择性的,如底物4i所示,其中在24小时后仅观察到起始材料向氟化产物的低转 化率(条目9)。最后,为了探索除苄基取代之外的反应性,使脂肪 族底物4j反应,得到6.8:1的取代/消除产物比,其中5j以54%的收率形成,以及回收的起始原料(条目10)。
为了解释上述反应性,DFT计算了苄基氟化过渡态TS1,其中吉布斯自由化能()为18.7kcal mol-1,由复合物1#39;F-和基质4a引起(方案1)。该过渡态结构的显着特征包括C-F键形成距离为2.1Aring;,C-Br键断裂距离为2.5Aring;。此外,在哈蒙德假设中,TS1代表了早期的过渡状态30 由计算的Wiberg键指数支持,对于断裂C-Br键为0.5,对于形成C-F键为0.2。从TS1 发现产物5a的形成以及复合物1bull;F- 的再生是一个用力过程。
a基于 1 H NMR计算收率。
3.结论
在扩展环丙烯离子作为相转移催化剂的用途中,已经报道了使用廉价的金属氟化物CsF的提供有机氟的苄基氟化反应方法。 1H, 19F NMR和UV-Vis光谱以及计算研究已经鉴定了原位衍生的环丙烯氟化物络合物作为该反应方法中的中间体。值得注意的是,这些复合物起源于弱但非常显着的非共价相互作用。这项工作的机械结果提供了可转移到广泛领域的见解,例如,不对称催化,生物工程和主客体分子离子传感。因此,我们实验室正在进行的努力旨在扩展环丙烯介导的相转移催化的所有组成部分。
4.实验部分
4.1.计算方法
使用密度泛函WB97XD和6-311 G(d,p)/ def2TZV基组进行计算31 使用高斯0932 并使用高斯视图v5.0.8可视化输出。为了解释溶剂效应,积分方程式形式极化连续溶剂化模型(IEFPCM)33在整个计算中使用。仅通过实际振动频率确认所有最小值。
4.2.材料和方法
材料从商业供应商获得,除非另有说明,否则无需进一步纯化即可使用。在高真空下加热至65℃干燥氟化铯(CsF)。所有反应均在惰性气氛(N2)下进行,并使用硅胶60F254通过薄层色谱(TLC)监测。在硅胶(230-400目)上进行快速柱色谱。除非另有说明,NMR光谱在CDCl3中的300MHz光谱仪(1H 300MHz,13C 75.5MHz,19F 292.4MHz)上记录。观察到的化学位移报告为相对于四甲基硅烷
(TMS)的d值(ppm)。四氯代环丙烯,34环丙烯氯35和苄基甲苯磺酸酯衍生物36 根据文献程序制备,并相应地匹配光谱。
4.3.环丙烯醛催化的一般程序
用CsF氟化苄基底物(5a-j)
向干燥的RBF中加入1·Cl-(21 mg,0.15 mmol),4-硝基苄基溴(85 mg,0.5 mmol)和CsF(227 mg,1.5mmol)并溶于丙腈(1mL)中。将所得混合物在回流下加热至65 ℃,持续7小时。除去溶剂,通过快速色谱法(己烷至乙酸乙酯= 6:1)纯化产物,得到5b,为黄色油状物。1 H NMR(300MHz,CDCl3,25℃)
全文共13393字,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[452728],资料为PDF文档或Word文档,PDF文档可免费转换为Word
您可能感兴趣的文章
- 通过对奥美拉唑合成反应的监测和定量反应的在线拉曼光谱和表征组件外文翻译资料
- 无金属碳基催化剂的研究进展外文翻译资料
- 钼酸钙/碳三维复合材料可控设计合成的研究外文翻译资料
- 生物催化选择性合成功能化喹唑啉酮衍生物外文翻译资料
- 三元V Zr Al ON氧氮化物-3-甲基吡啶氨氧化的高效催化剂外文翻译资料
- 综述纳米零价铁(nZVI)的合成,特性和在环境修复中的应用外文翻译资料
- 自消毒PVC表面使用点击化学设计外文翻译资料
- 微波辅助直接合成4H-1,2,4-苯并噻二嗪1,1-二氧化物衍生品外文翻译资料
- 微波辅助下直接合成1,1-二氧代-4H-1,2,4-苯并噻二嗪类衍生物外文翻译资料
- 压力选择在变压精馏中的重要性外文翻译资料

